������ ��ϸ�л��ϳɵĹ���ѧ����
3.1 �� ��
��ϸ�л��ϳɵĹ���ѧ��Ҫ�����������ݣ��Ծ����Ʒѡ���ȷ���ڼ����Ϻ;�����������ĺϳ�·�ߺ���·�ߣ��Ե�Ԫ��Ӧȷ����ѹ����������ϳɼ�������ɷ�Ӧ�ķ������Եõ��������������ʵIJ�Ʒ��
��ν�ϳ�·�ߣ�ָ����ѡ��ʲôԭ�ϣ������ļ�����Ԫ��Ӧ���Ʊ�Ŀ�IJ�Ʒ�����磬��1.6���ᵽ�����ӵ����������кü����ϳ�·�ߣ����Ǹ�����ȱ�㡣���ںϳ�·�߽���Ͼ����Ʒ�ڸ���Ԫ��Ӧ�����ۡ�
��ν����·�ߣ�ָ���Ƕ�ԭ�ϵ�Ԥ�������ᴿ�����顢����ۻ����ܽ⡢���������������ȡ���ȴ�ȣ��ͷ�Ӧ��ĺ��������������ա���������ȡ���ᾧ�����������ˡ�����ȣ�Ӧ������Щ�������̣���Ԫ��������ʲô�豸��ʲô�������̵ȡ�
��ν��Ӧ����ָ���ǣ���Ӧ��ķ��ӱȡ���Ҫ��Ӧ���ת���ʣ���Ӧ��ȣ�����Ӧ���Ũ�ȡ���Ӧ���̵��¶ȡ�ʱ���ѹ���Լ���Ӧ����������Ӧ�����������ܼ���ʹ�ú�ѡ��ȡ�
��ν�ϳɼ�����Ҫָ���ǣ��Ǿ���Ӵ�������ת�ƴ���������ϴ������л��ϳɺ͵���л��ϳ��Լ�ø���ȡ�
��ν��ɷ�Ӧ�ķ�����Ҫָ���ǣ���Ъ����������������ѡ��Ӧ����ѡ�����Ƶȡ�Ϊ����ɻ������������DZ�������漰�����ϵ������г���˽⡣�������ϵ���Ҫ������Ҫ�У�
��1��������һ�������µĻ�ѧ�ȶ��ԡ����ȶ��ԡ����ȶ����Լ������ȶ��ԣ������������ˮ�ֳ��ڽӴ����ȶ��ԣ��ȡ�
��2���۵㣨���̵㣩���е㡢�ڲ�ͬ�¶��µ�����ѹ��������ˮ�е��ܽ�ȡ�ˮ��Һ̬�����е��ܽ�ȣ�������ˮ�Ƿ��γɺ����Լ�����¶Ⱥͺ������ɵȡ�
��3�����ء��۹��ʡ����ȡ�����ϵ���������ȡ��ӷ��Ժ�ճ�ȵȡ�
��4�����㡢��ը���ͱ�Ҫ�İ�ȫ��ʩ��
��5�����ԡ��������Σ�����ڿ����е�����Ũ�ȡ���Ҫ�ķ�����ʩ�Լ��ж��ļ��ȴ�ʩ��
��6�����ϵ���Ʒ��������ʺ����Ӽ��������������۸�Ӧ��Դ����װ�ʹ���Ҫ��ȡ�
�������ʿ��Բ��ĸ����й��Ӳᡣ
3.2.1 ��Ӧ���Ħ����
��Ӧ���Ħ����ָ���Ǽ��뷴Ӧ���еļ��ַ�Ӧ��֮���Ħ����֮�ȡ����Ħ���ȿ��Ժͻ�ѧ��Ӧʽ��Ħ����֮����ͬ�����൱�ڻ�ѧ�����ȡ����Ƕ��ڴ�����л���Ӧ��˵��Ͷ�ϵĸ��ַ�Ӧ���Ħ���Ȳ������ڻ�ѧ�����ȡ�
3.2.2 ���Ʒ�Ӧ�������Ӧ��
��ѧ��Ӧ�ﲻ����ѧ������Ͷ��ʱ����������С��ѧ���������ڵķ�Ӧ����������Ʒ�Ӧ�����ij�ַ�Ӧ������������Ʒ�Ӧ����ȫ��Ӧ������������÷�Ӧ���Ϊ��������Ӧ���
3.2.3 �����ٷ���
������Ӧ�ﳬ�����Ʒ�Ӧ����������������ռ�����������İٷ��������������ٷ�����������Ne��ʾ������Ӧ���Ħ������Ni��ʾ�������Ʒ�Ӧ����ȫ��Ӧ�����ĵ�Ħ������������ٷ���Ϊ��
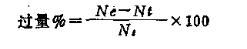
3.2.4 ת���ʣ���x��ʾ��
ijһ�ַ�Ӧ��A��Ӧ����ռ����Ӧ�����������İٷ���������Ӧ��A��ת���ʡ�
3.2.5 ѡ���ԣ���s��ʾ��
ѡ����ָ����ijһ��Ӧ��ת���Ŀ�IJ����������ĵ�Ħ����ռ�÷�Ӧ���ڷ�Ӧ��ʵ�����ĵ�����Ħ�����İٷ������跴Ӧ��A����Ŀ�IJ���P��Np��ʾ����Ŀ�IJ����Ħ������a��p�ֱ�Ϊ��Ӧ��A��Ŀ�IJ���P�Ļ�ѧ����ϵЧ����ѡ����Ϊ��
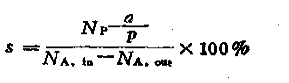
3.2.6 �������ʣ���y��ʾ��
����ָ�������ɵ�Ŀ�IJ����Ħ����ռ����ķ�Ӧ����Ħ�����İٷ�������������ֽ����������ʡ�
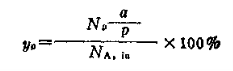
ת���ʡ�ѡ���Ժ�������������֮��Ĺ�ϵ�ǣ�y=s��x��
3.3 ��ѧ��Ӧ��
3.3.1 ����
��ѧ��Ӧ���Ƿ�Ӧԭ�������н��л�ѧ��Ӧ������Ŀ�IJ�����豸����ѧ��Ӧ���ڽṹ�ϺͲ����ϱ�����������Ҫ��
��1�� �Է�Ӧ���ϣ��ر��ǷǾ������Һ��Ӧ�ҺҺ��Ӧ����̷�Ӧ�Һ�̷�Ӧ���Һ�����෴Ӧ��ṩ���õĴ������������ڿ��Ʒ�Ӧ��ϵ��Ũ�ȷֲ���������Ŀ�ķ�Ӧ��˳�����С�
��2�� �Է�Ӧ���ϣ��ر���ǿ�ҷ��Ȼ�ǿ�����ȵķ�Ӧ��ϵ���ṩ���õĴ���������������ЧӦ���Ƴ����������ڷ�Ӧ��ϵ���¶ȿ��ơ�
��3���ڷ�Ӧ���¶ȡ�ѹ���ͽ��������£��������õĻ�еǿ�Ⱥ���ʴ���ܡ�
��4�� ����Ӧ��Ӧ���IJ�����ʽ����Ъ������������������
3.3.2 ����������������
�ڷ�Ӧ����ʵ��һ����ѧ��Ӧ���������ֲ�����ʽ������Ъ������������������Ъ�����ǽ����ַ�Ӧԭ�ϰ�һ����˳��ӵ���Ӧ���У�����һ�����¶ȡ�ѹ���¾���һ��ʱ������ض��ķ�Ӧ��Ȼ��Ӧ�õ����ϴӷ�Ӧ���зų�����Ϊ��Ӧԭ���Ƿ����ӵ���Ӧ���еģ������ֽ������������������ڼ�Ъ����ʱ����Ӧ��������ʱ����ı䡣���⣬��Ӧ����¶Ⱥ�ѹ��Ҳ������ʱ����ı䡣���������ǽ����ַ�Ӧԭ�ϰ�һ���ı����ͺ㶨���ٶ��������ϵؼ��뵽��Ӧ���У����Ҵӷ�Ӧ�����Ժ㶨���ٶ��������ϵ��ų���Ӧ��������������£���Ӧ����ijһ�ض���λ�ķ�Ӧ���ϵ���ɡ��¶Ⱥ�ѹ��ԭ�����Ǻ㶨�ġ�
���������ȼ�Ъ�����������ŵ㡣��һ�����������Ƚ�����ʵ�ָ߶��Զ����ƣ���Ʒ�����ȶ�������Ъ�����ij����Զ��������൱���Ѷ��ҷ��ð�����˼�Ъ����������������Ҫ�϶���Ͷ������ҷ�Ӧ��Ч���������˵�����Ӱ�졣�ڶ�����������������ʵ�����̷�Ӧʱ�䡣����Ъ��������Ҫ�м��ϣ������������¶Ⱥ�ѹ�������ϣ��Լ�����һ��Ͷ�ϵȸ�������ʱ�䡣��ˣ�����������ģ��Ӧʱ��̵Ļ�ѧ���̶������ܲ��������������ر������෴Ӧ��������Ӵ�����Ӧ������������������������������������ʵ�ֽ��ܡ�����ӷ�Ӧ�����Ƴ��������Լ��ȵķ�Ӧ��������ȴʱͨ���Ƚ�����������������������Ԥ����ķ�Ӧԭ�ϣ����߰��������ݸ�ˮ�Բ���ˮ��������Ҫ�Ѽ�Ъ������ϵ�����ϵͳ��һ��������ʵ�ֵġ�
���ǣ���Ъ����Ҳ�������ص��ŵ㡣��һ�����������ļ��������ȼ�Ъ�������ѵöࡣ���ܺͽ�ʡ�Ͷ���һ������������ģ���������ģ�����С��ģ������˵������һ���������������Dz�ֵ�õġ��ڶ�����Ъ�����Ŀ�����ͣ��һ��������������ף���Ъ�������豸�ڲ����Ĵ�С���н϶��������أ�������ƷҲ������ԣ��������������豸ͨ��ֻ��������һ��Ʒ����������ijЩ����£����ڷ�Ӧԭ�ϻ������������ʣ�����ճ���Ⱥͷ�ɢ״̬�����������ڷ�Ӧ�����Ŀ��ƣ������¶ȡ�ѹ���Ͳ������裩�����أ����ڲ������������������ù��෨����2,3-��ͱ�����ü�Ъ��������ˣ����ڶ�Ʒ�֡�����С�ľ�ϸ������Ʒ��˵����Ъ�������൱�㷺��Ӧ�á�
3.3.3 ��Ъ������Ӧ��
ҺҺ���Һ�����Ъ�����ķ�Ӧ�������Ϻ�ʵ�鱦���豸���ƣ�����ͬ���ǹ�ģ����������ഫ�ȷ�ʽ��ͬ�����ּ�Ъ������Ӧ�������dz��ڵķ�Ӧ�ۣ��൱���ձ�����Ҳ�����Ǵ������������ķ�Ӧ�����൱���Ŀ���ƿ������������ѹ�ĸ�ѹ��������ijЩ���Ϸdz�ճ����Һ���෴Ӧ����������ʽתͲ��ĥ��ʽ��Ӧ��(�����)����õĴ��ȷ�ʽ���ڹ��ⰲװ�����ڹ��ڰ�װ�����̹ܡ���ȴһ������ˮ���䶳��ˮ������һ����ˮ��������Ҫ�ϸ��¶�ʱ��130~260�棩�������µ����͡�����תͲ��ĥ��ʽ��Ӧ�������籽�����౺��ǻ��ƶ��������ᣬ����Լ200�棩���������·�Ӧ���̣�����2-�������Ƶļ�����2-���ӣ�����28~320�棩�������Բ���ֱ�ӻ���ȣ�ȼ��ú����ȼ���ͻ�ú������ֱ���õ��ȡ��ڸ�������£�Ҳ����ֱ����Ӧ���м������������ȴ�������ص�����ż�Ϸ�Ӧ��������ֱ��ͨ��ˮ�������м��ȣ�����ijЩ��ϵ�����ˮ�ⷴӦ����
3.3.4 Һ��������Ӧ��
�����������ķ�Ӧ���У������ּ�������ģ�ͣ������������͡��͡������û��͡���
3.3.4.1 �������ͷ�Ӧ��
��ͨ����װ�н������ʹ���װ�õķ�Ӧ������ͼ��ʾ��
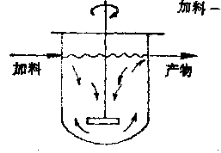
��Ӧԭ���������ϵؼ��뵽���У��ڽ������ڹ���ͣ��һ��ʱ�䣬ͬʱ��Ӧ����Ҳ�������ϵشӹ������������ַ�Ӧ�����ص��ǣ�ǿ�ҵĽ�������˷��������ã���Ʒ��죩�����¼�����������Ѵ��ڹ��ڵ�������˲����ȫ��ϣ����Թ��ڸ������ϵ���ɺ��¶ȶ���ͬ�����ҵ��ڳ��ڴ����ϵ���ɺ��¶ȡ����ǣ������и��������ڷ�Ӧ���ڵ�ͣ��ʱ������ͬ��
��ʽ������Ӧ������Ҫ�ŵ���ǿ�ҵĽ��������ڷǾ��෴Ӧ��Ĵ��ʣ��ɼӿ췴Ӧ�ٶȡ����⣬Ҳ������ǿ�ҷ��ȷ�Ӧ�Ĵ��ȣ��ɼӴ�Ӧ�����������������磬�����ױ����ȱ���һ���������ù�ʽ������Ӧ����
��ʽ��������Ҳ�кܶ�ȱ�㡣��һ���������ϵ���ɵ��ڳ������ϵ���ɣ������з�Ӧԭ�ϵ�Ũ���൱�ͣ��������Ӱ�췴Ӧ�ٶȡ��ڶ��������ķ�Ӧ������Ҳ�Ʊز�����һ������δ��Ӧ��ԭ�ϴӶ�Ӱ�����ʡ������������Ѿ����ɵķ�Ӧ�����Ũ���൱�ߣ�������һ��������������Ӧ�����磬���û����һ�������̣�������õ������������������豸���������ͣ���Ӧ�����к��н϶�δ��Ӧ�ı������ᣬ������Dz�Ʒ�������к��иߴ�2~4%�ĸ����������������ˣ��������������ڹ�ҵ�Ϻ��ٲ��á�
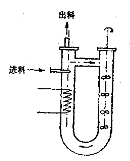
Ϊ�˿˷����ȱ�㣬һ�㶼���ö��������������ͼ��ʾ��
��Ӧԭ�������ļӵ���һ����Ӧ���У���Ӧ�������ε����������ڶ������͵���������Ӧ��������Ӧ����������һ����Ӧ��������������������������ص��ǣ�������Ӧ��֮�䲢Ԫ�������ã��Ӷ�����˷������õIJ���Ӱ�졣��������������ŵ㣺��һ����Ӧ���з�Ӧԭ�ϵ�Ũ�ȱȽϸߣ���Ӧ�ٶ��൱�죬�ɴ������豸������������ÿ����Ӧ�ÿ��Կ��Ʋ�ͬ�ķ�Ӧ�¶ȣ����⣬�����һ����Ӧ���У���Ӧԭ�ϵ�Ũ���Ѿ���úܵͣ��������ٷ�Ӧ������ʣ��δ��Ӧ��ĺ����������ڽ���ԭ�ϵ����Ķ����������������Ӧ���������������Ϊ�˱���ԭ���Զ�·�ķ�ʽ�Ӹ���Ӧ���������������ڹ��ڰ�װ����Ͳ�����ǽ������߹����ɵ��������ʽ����ͼ��ʾ��
����Ҳ���ѷ�Ӧ������U��ѭ���ܵ���ʽ����ͼ��ʾ��
3.3.5 ��Һ�෴Ӧ��
��Һ�෴Ӧ��Ҫ�����ÿ���������������Ӧ�������������Ȼ���Ӧ�Լ��������ļ��ⷴӦ�ȡ����ģ������һ�������ù�����ʽ��Ӧ�������෴Ӧ���ȿ��Լ�Ъ������Ҳ������������������������ʱ,��Ӧ�������Ǵ����ĵײ����룬β�������Ķ����ų�����Һ̬���ϼ����Դ����ĵײ����룬�������ϲ�����������������Ҳ���Դ������ϲ����룬�����ĵײ�������������������ͼ��ʾ��

��Һ����ʽ��Ӧ��
��Ϊ��������һ���ķ��������ã�Ϊ�˼������IJ���Ӱ�죬��ʹ��Һ��֮�������õĴ������ã���������װ�����ϡ�ɸ�塢���ְ塢���ֵ�������ڲ�������Ϊ�˿��Ʒ�Ӧ�¶ȣ��ɲ����ڲ��Ƚ���������ѭ��ʽ�Ƚ�������Ϊ�˱�������̫�߶�����ͨ�������ѹͷ��Ҳ�������ö��������ķ�ʽ����ÿ�����ĵײ�ͨ�뷴Ӧ���塣
����Һ�෴Ӧ�ٶ��൱��,��ЧӦ�൱��ʱ��Ҳ���Բ����й�ʽ������Ӧ���������̼�������Ȼ�����Ĥ������ʽ��Ӧ��������ʮ��������ǻ�����
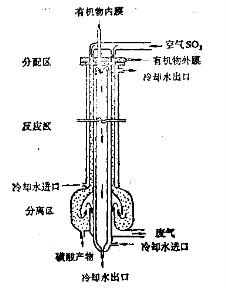
˫Ĥ��Ӧ��
3.3.6 ������Ӵ�����Ӧ��
������Ӵ�����Ӧ�ǽ���Ӧԭ�ϵ���̬�������һ�����¶ȡ�ѹ����ͨ�������������ɵġ����෴Ӧ��ʽ�ڹ�ҵ���й㷺��Ӧ�ã���һ�㶼�������������ķ�ʽ�����෴Ӧ����Ƶ���Ҫ�����Ǵ��Ⱥʹ�����װж�����෴Ӧ����Ҫ���������ͣ������ȹ̶�����Ӧ�����й��Ŷ�����Ӧ������������Ӧ����
3.3.6.1 ���ȹ̶�����Ӧ��
������ȹ̶�����Ӧ���Ľṹ�dz�������ͼ��ʾ������һ��û�д���װ�ã�ֻװ�й����������������Ӧԭ�ϴ�������һ�����룬��Ӧ���������������һ����������෴Ӧ������Ҫ�ŵ����豸�ṹ���ռ������ʸߡ���۵͡�����װж���ס����ǣ������෴Ӧ���У���Ӧ���Ϻʹ������¶��DZ仯�ġ����ڷ��ȷ�Ӧ���ӽ��ڵ������¶������ߡ��������ȷ�Ӧ���ӽ��ڵ������¶����͡����ҷ�Ӧ���̵���ЧӦԽ�����ڵ��²�Խ����������ص�ʹ�õ�����ȹ̶�����Ӧ��ֻ�����ڹ�����ЧӦ����Ӧ����Ƚ��ȶ����Է�Ӧ�¶ȱ仯��̫���С���Ӧ�����º����к��д����������壨����ˮ������������һ��ͨ����Ӧ��ת���ʲ�̫�ߵĹ��̡����磬�ȱ�������ˮ���Ʊ��ӣ��״������������Ƽ�ȩ�ȣ����⣬������ȹ̶�����Ӧ���еĴ����㲻��̫��������ںͳ��ڵ��²�̫����ˣ�ֻ�����ڷ�Ӧͣ��ʱ��̵Ĺ��̡�

������ȷ�Ӧ��
����Ӧ����ЧӦ�ϴ�ʱ��Ϊ�˸��Ʒ�Ӧ���¶����������ת���ʣ��������ö�ξ��ȷ�Ӧ��,��ͼ��ʾ��
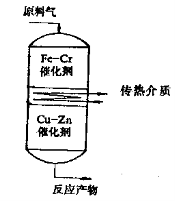
COˮ�����任��Ӧװ��ʾ��ͼ
Ϊ�˵�����Ӧ�¶ȣ��ɸ��ݹ��̵��ص㣬ѡ����ʵ��������ȴ�������ڷ��ȷ�Ӧ���ɽ���ԭ������Ԥ�ȣ����磬һ����̼��ˮ����ת�����⣩���������ȷ�Ӧ�����Բ����ⲿ��ʽ����¯��
3.3.6.2 �й�ʽ�̶�����Ӧ��
�й�ʽ�̶�����Ӧ���Ľṹ���ͺܶ�,��Ľṹ�����ڵ����й�ʽ����������ͼ��ʾ��
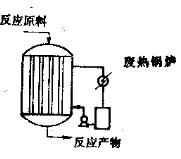
�й�ʽ�̶�����Ӧ��
���������й��ڣ��������ڹ��������ȴ����ȡ����ڷ��ȷ�Ӧ�����������λ����������彫��ЧӦ�Ƴ����ȵ������徭���ȹ�¯���º��ٷ����йܷ�Ӧ�������ȹ�¯����������ɲ���0.6~2.0MPa��ˮ�����������ǵȷ��ӱ�����غ��������ƵĻ����۵�141�棬����147~540������������ŵ��DZ��Ⱥ͵���ϵ������Ч���á��������ȷ�Ӧ��������Ҫ����¶ȣ�������Һ̬������̬����������м��ȡ�����Ҳ�����õ��Ȼ����ⲿ��ʽ¯���ȣ���ι̶�����Ӧ������
3.3.6.3 ��������Ӧ��
���Ļ����ṹ��ͼ��ʾ����Ҫ�����ǿ��塢����ֲ��塢�Ƚ���������������װ�á���ʱΪ�˼��ٷ����ϲ�������̬�����������ڴ��������ڸ��ӵ���������ڲ�����
��̬���Ļ���ԭ���ǣ������徭���ֲ������ʵ��ٶȾ��ȵ�ͨ����״��������ʱ�������Ŀ�����������Ư������������������ļ����˶��������������Ʒ��ڵ�Һ��һ�����ܹ������˶��������ֽ������ڴ���
����������Ҫ�ŵ��ǣ�����ϸ�������������ڷ�Ӧ�����ڴ������е�����ɢ����������������ʸߡ���ǿ����Ĵ��ȣ������¶Ⱦ��ȣ��ɿ�����1~3����¶ȲΧ�ڡ����ڴ���������������������ñ��й�ʽ�̶����͵öࡣ��������Ӧ���㷺���ڿ������������ѻ��ȷ�Ӧ���̡�
����������Ҫȱ���ǣ����ڷ������ã�����ijЩ��Ӧת���ʺ�ѡ���Բ���̶�������������ĥ����ʧ������ʹ�ñ����Ϳ���״������
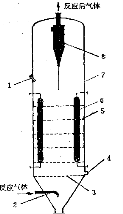
��������Ӧ��
1-�Ӵ����ڣ�2-Ԥ�ֲ�����3-�ֲ��壬4-ж�����ڣ�
5-�ڲ�������6-�Ƚ�������7-���壬8-���������
3.4 ��ϸ�л��ϳ��е��ܼ�ЧӦ
3.4.1 ����
3.4.1.1 �ܼ����л���Ӧ��Ӱ��
�ܼ������ò�ֻ��ʹ��Ӧ���ܽ⣬����Ҫ�����ܼ����Ժͷ�Ӧ�����������á����ѡ����ʵ��ܼ��Ϳ���ʹ����Ӧ�����ؼ��٣���������Ч�����Ƹ���Ӧ�����⣬�ܼ�����Ӱ�췴Ӧ���̡���Ӧ���������廯ѧ����ˣ��˽��ܼ������ʡ������Լ��ܼ�������֮�������ã���������ѡ���ܼ�������Ŀ�ķ�Ӧ��˳���������Ҫ���塣
3.4.1.2 ��Һ���ܽ�����
�����ܽ����ܼ����γɵľ�̬�������ϵ������Һ���ܽ����õ�����ϵľ�������ǡ��������ܡ����ܵ���˵��һ�����������ڻ�ѧ�ṹ���Ƶ��ܼ������������ڻ�ѧ�ṹ��ȫ��ͬ���ܼ����������������ڼ����ܼ����Ǽ������������ڷǼ����ܼ������ǣ�Ҳ��һЩ���⣬����ѧ�ṹ���Ƶ���ֳ��ֲ����ԣ�����ѧ�ṹ�����Ƶ����ȴ�ܻ����ܽ⡣һ����Ϊ���ܽ������йص�������Ҫ�У�
��1����ͬ����֮��������벻ͬ����֮������������ϵ��
��2���ɷ��ӵļ���������ĵϳ̶ȡ�
��3���ܼ������á�
��4���ܼ������ʵķ�������
��5���ܼ����Ի��ŵ��������Ŀ
3.4.1.3 �ܼ�������֮����������
�����ܼ����Ӻ��������ʷ���֮�������������Է�Ϊ�����ࣺ
��1�������� ������������������������-������������-ż������
��2�����»���Van der Waals���� ����ھ�����������ż����ż����(������)��ż��-�յ�ż�������յ�������˲ʱż�����յ�ż������ɫɢ������
��3��רһ���� ��������������á����ӶԸ���/���Ӷ���������ã����ת���������ܼ������á����ӻ����á�������ú����ܼ�����õȡ�
��һ��͵ڶ�����Ӽ�������ͨ���ڵķ�רһ��������������Ӽ�����ֻ����һ���ṹ֮����ܷ����ġ���һ�������רһ������
3.4.2 �ܼ��ķ���
�ܼ��ķ����������������һ������;��
3.4.2.1 �ܼ�����ѧ�ṹ����
�ܼ�����ѧ�ṹ���Է�Ϊ���ܼ����л��ܼ������ࡣ���õ����ܼ��������١����ܼ�����ѧ�ṹ���࣬���Ը���ijЩ���Ե�Ԥʾ�������ǰ���ᵽ�ġ��������ܡ�ԭ�����⣬���ݸ����ܼ���ѧ��Ӧ�Ե�֪ʶ��Ҳ���������Ǻ�����ѡ���ܼ������������ʺ��ܼ�֮�䷢����ϣ���ĸ���Ӧ��
3.4.2.2 �ܼ���ż�������ͽ�糣��������
ż�������ͽ�糣�����DZ�ʾ�ܼ����Ե�������Ҫ������������ַ��෨������Ҫʵ�����塣
1. ż������
�����о�������ż���ص��ܼ����������ԡ�������֮��������û������ż���ص��ܼ�����������ԡ��Ǽ��ԡ��ܼ������缺�顢�����顢�������Ȼ�̼�Ͷ���̼�ȡ�����û������ż���̵��ܼ��Ǽ��ٵģ���˰�ż����С��2.5D�ķ������������ܼ��������ȱ��Ͷ������飩Ҳ��Ϊ�Ǽ����ܼ���
ż������ҪӰ�������ʣ����ӻ����ӣ���Χ���ܼ����ӵĶ������á�
2. ��糣����
����ܼ����ӱ���û������ż���أ�����糡��ʹ�ܼ������ڲ��������ɶ������յ�ż������������ż�����յ�ż�����ܼ����ӱ����ĵ�������ǿ�Ƶ��γ�һ���������У��Ӷ�������ν�ġ��������á�����������Խ��,�糡ǿ�ȵ��½�ҲԽ��EֵԽС����糣����Խ����ˣ���糣����ʾ�ܼ����ӱ����������ɵ����������ܼ�ʹ����ż�������������
3. �ܼ����Եı������ܼ�������
�����ܼ��ġ����ԡ�������ֱ��������δ��ȷ�е��¶��塣�����ܼ�����ν���ԣ���Ҫ���������ܵ��ܼ����������ܼ�������ָ����ÿһ�����ܽ�ķ��ӣ������ӣ���һ����ܼ����ӻ��ɻ���ذ�Χ�������ܼ���������һ��ʮ�ָ��ӵ������������ܼ�������֮������רһ�Ժͷ�רһ������õ��ܺ͡���������ܼ�����������ã�������һ��������������ʾ��ϰ���ϣ�������ż���ػ��糣����ʾ��������������������ֻ�ܷ�ӳ�ܼ���һ�������ʻ���ij�����á�������ˣ����ܼ��������ǵļ��ԣ�ż���ػ��糣�����ֳɼ��ֲ�ͬ�����ͣ����ֱ����۸����ܼ������ʷ��ӣ������ӣ������ã�����Ϊ���־��巴Ӧѡ����ʵ��ܼ���������Ҫʵ������ġ�
3.4. 3 �л���Ӧ���ܼ���ʹ�ú�ѡ��
3.4.3.1 �л���Ӧ���ܼ���Ҫ��
���л���Ӧ���ܼ���ʹ�ú�ѡ�������˿����ܼ�������Ӧ���ٶȡ���Ӧ���̡���Ӧ�������廯ѧ��Ӱ�����⣬�����뿼���������أ�
��1�� �ܼ��Է�Ӧ��ͷ�Ӧ���ﲻ������ѧ��Ӧ����Ӱ������Ļ��ԡ��ܼ������ڷ�Ӧ�����¼��������������ȶ��ԡ�
��2�� �ܼ��Է�Ӧ���нϺõ��ܽ��ԣ�����ʹ��Ӧ�����ܼ��������÷�ɢ.
��3�� �ܼ����״ӷ�Ӧ���л��գ���ʧ�٣���Ӱ���Ʒ��������
��4�� ���ܼ������ܲ���Ҫ̫�ߵļ�����ȫ��ʩ��
��5�� �ܼ��Ķ���С�����ܼ��ķ�ˮ���״�����
��6�� �ܼ��ļ۸���ˡ���Ӧ���㡣
3.4. 3.2 ���෴Ӧ�������ܼ�
1. ����
�������������ȼ��顢�������飻
ϡ�����������ȱ����ڶ��ȱ���
����������Ũ���ᡢ���ᡢ����Ũ���
2. �ǻ�
�ܼ��ǻ������ᡢ���ȼ��顢���Ȼ�̼
����������Ȼ���ǻ���1,2-�������顢1,1,1-�������顢�ڶ��ȱ������ȱ�����������
����ǻ����ڶ��ȱ������ȱ�
���ȥˮ�ǻ����ڶ��ȱ���ú��
3. ±��
��ˮ���ʣ�Ũ���ᡢ�Ȼ��ᡢ���Ȼ��ס��������ס����Ȼ��ѡ����Ȼ�̼���������顢�ȱ����ڶ��ȱ�������
ˮ���ʣ��ȱ����ڶ��ȱ�����������
4. ������
��̼�����������������ᡢ��ͪ��ˮ����������������
5. ����
Ũ���ᡢ���ᡢˮ������������ʯ���ѡ������������
6. Friedel-Crafts��Ӧ
�ܼ������������顢�������顢1,1,1-�������顢����̼��ʯ���ѡ���������������Һ̬��Ӧ���
���ڷ���ALCl3-NaCl��AlCl3-NaCl-H2NCONH2
3.5 ������Ӵ���
3.5.1 ����
������Ӵ�����Ӧ�ǽ���̬��Ӧ����һ�����¶ȡ�ѹ����������ͨ����������ı������ɵġ����ַ�Ӧ��ʽ��Ӧ�������Ԫ��Ӧ��
�������ͨ��������Ҫ���������ʡ�����������������ɡ���ʱΪ�˱����Ƴ�����Ҫ����״����ƴ����Ļ�еǿ�Ȼ��϶�ṹ�����Ʊ�����ʱ��������ͼ���������ʡ�
��������������ȿ��Է�Ϊ����״�ͷ�ĩ״�������͡�����״�������ڹ̶�����Ӧ������ĩ״����������������Ӧ����
����������ձ�����ֿ��Է�Ϊ�߱ȱ����ͺ͵ͱȱ��������ࡣ�����ı�����������Ϳ�϶�е��ڱ��������֡�ÿ�˴������ܱ���������ȱ���,���ĵ�λ��m2/g��
����������ܶȣ�s/mL�����ӱ�������ʾ�����ǰ�һ�������Ĵ���������Ͳ�У�ֱ�ӹ۲����������õġ�
���ڹ�����������ã���Ȼ�Ѿ�������ٴ����ۣ����ǻ�û��һ��������ȫ��ء����Ƶؽ������и��ֽӴ�����Ӧ�Ļ�������õ������ǻ����������ۡ�������ѧ˵�Ͷ�λ�������ѧ˵�ȡ���Щѧ˵��Ҫ���Ǵ����ı���ֻ��һС�����ض��IJ�λ��������ã���Щ��λ�����������ġ���Ӧ����ӵ��ض������ڻ������ķ�����ѧ�������γɻ����Ȼ�������������һ������һ��δ�������ķ�Ӧ����������ã�����Ŀ�IJ�����������ַ�Ӧ����ӷֱ��������ڵIJ�ͬ�Ļ��������������ֱ����ɻ����Ȼ����������������ö�����Ŀ�IJ�����ڻ������ĵ������ԣ�����һ�������Ĵ�������ֻ��ijһ������ijһ�����巴Ӧ�������õĴ����ã�����Ŀ�ķ�Ӧ�������õ�ѡ���ԡ�
3.5.2 ������ѡ���ԡ����Ժ�����
3.5.2.1 ������ѡ����
��ָ�����ض�����ר�Ŷ�ijһ��ѧ��Ӧ��������õ����ܡ���ѡ����Ҳ����ijһ��Ӧ��ͨ��������ת��ΪĿ�IJ���ʱ�������ĵ�Ħ����ռ�÷�Ӧ���ڷ�Ӧ��ʵ�����ĵ�����Ħ�����ٷ�������ʾ��������ѡ�������������ɡ��Ʒ��ͷ�Ӧ�����������йء�
3.5.2.2 �����Ļ���
�ڹ�ҵ�ϣ������Ļ���ͨ���õ�λ�������λ�������������ض���Ӧ�����£��ڵ�λʱ�������õ���Ŀ�IJ������������ʾ������ijЩ����Ӧ����ҵ�����Ļ��Ի�ʹ�����ض�������ʱ���ٶ��£���Ӧ���ת���ʻ�Ŀ�IJ������������ʾ��
3.5.2.3 ����������
��ָ���Ǵ����ڹ�ҵ��Ӧ����ʹ�õ���ʱ�䡣������ʹ�ù����У������¶ȡ�ѹ�������ա������Ӱ�죬�Լ����ͻ��̼�����ɵ���������������ٵ�ʹ��������ijЩ�����Ļ�ѧ�ı仯�������۽ᡢ�ۻ��Լ��ᾧ�ṹ��ȱ���ı仯�ȣ���Щ����Ӱ������еĻ������ģ��Ӷ�Ӱ������Ļ��Ժ�ѡ���ԡ��������Ļ��Ժ�ѡ�����½���һ���̶ȣ����Ҳ����跨�ָ������ʱ������Ҫ������������ҵ�����������뷴Ӧ���͡���������ɺ��Ʒ��������йء���Щ�����������ɳ������꣬�еĴ�������ֻ�м�Сʱ��
����ʹ��һ��ʱ���������½�����Ҫ����������ʹ��ʱ����������Ļ���ڡ�
3.5.3 ���������
3.5.3.1 ����������
��ָ���Ƕ�Ŀ�ķ�Ӧ�������ô����Եijɷ֡����ھ��巴Ӧ���������������ͨ������ʵ��ɸѡ�����ġ���ͨ���ǵ�һ�ɷֻ�������ֳɷ֡����磬����ǿ������Ӧ�Ĵ�������������ͨ������������������
3.5.3.2 ������
���DZ���û�д����Ի�����Ժ�С��������ߴ��������ʵĻ��ԡ�ѡ���Ի��ȶ��Եijɷ֡��ڴ�����ͨ������������������������������Ҫ���ڸ������ȶ��ĸ��ֽ���������ǽ�������������κͽ���Ԫ�ء�
3.5.3.3 ����
�����Ǵ�������ֺ���������֧���ճ������ɢ�塣����ʹ�����塢�ڴ����д�������ֺ��������ĺ������Ժܵ͡�����Ļ�е���������Ӵ�������ֵıȱ��棬�������������Ӷ��ӳ�������������ʹ���������㹻�Ŀ�Ϯ�ȡ���еǿ�ȣ�Ӳ�ȡ���ĥ�ԡ���ѹǿ�ȵȣ������ȶ��ԡ����Ⱥ͵����ʵȡ����⣬��Щ���廹�������������ַ���ij�ֻ�ѧ���ã��ı��˴�������ֵĻ�ѧ��ɺͽṹ���Ӷ������˴����Ļ��Ժ�ѡ���ԡ���ˣ����Ʊ�����ʱ�����ѡ��Ҳ�Ǻ���Ҫ�ġ�
3.5.4 �����Ķ���ж�������
���������������ʵ�Ӱ�죬ʹ����Ժ�ѡ�����½�����������������ж������������ʽ��������Ķ��
3.5.4.1 �����Ķ���
�ڹ�ҵ�����У������Ķ���ͨ�����Է�Ӧԭ�ϡ���ʱ����Ҳ�������ڴ����Ʊ������л���ģ�����������������ȾԴ�������ж�����ͨ�������ڴ�������ֱ���Ļ��������ϣ������������������������������½���
3.5. 4.2 �������ж�
�ж������ڶ������������ַ�����ij�����ã�����ƻ����ڸ��˻�����������ɵġ������ڻ����������������Ͻ����������üķ���ʹ�����ָ����Ե��ж���������������ж�������ʱ�ж�����������������Ľ�Ϻ�ǿ��������һ�㷽���������ȥ���ж�����������������ж����������ж�����������ʱ�ж������跨���������������ж�����Ҫ�������ʴ�����
3.5.4.3 �����ж���Ԥ��������
Ϊ�˱���������ж���һ�����ʹ�����Ͷ������ʹ��ǰ����Ӧָ����Щ�Ƕ���Լ���Щ�����ڷ�Ӧԭ���е����������������ԭ�����к����ʵĺ��������涨ʱ�������ԭ�Ͻ��о��ƣ���������ԭ�ϡ�������ʱ�ж����跨�����������ķ���ͨ�����ÿ�����ˮ������������һ���¶���ͨ�������Գ�ȥ��̼�������������ȶ�������������½�������ʹ�ýϳ�ʱ�����Ҫ��������ʱ���������̿��Ծ��ڷ�Ӧ���н��С�
3.5.5 �������Ʊ�
һ�������Ĵ���һ��Ӧ�߱��������ܡ�
��1�� ���Ըߡ�ѡ���Ժá����ȺͶ����ȶ���ʹ��������������������
��2�� ��еǿ�Ⱥ͵����Ժá�
��3�� ���к��ʵĺ�۽ṹ�����磬�ȱ��桢��϶�ȡ����ֲ��������Ⱥ����ṹ�ȡ����ֺ�۽ṹ��Ҫ�ṩ�㹻�Ĵ����棬��Ҫ��ʹ��Ӧ��Ͳ����ڷ�Ӧ������˳����ɢ��
(4)�Ʊ���㡢�۸���ˡ�
���Ʊ�����ʱ������ʹ��һϵ�л�ѧ�ġ������ĺͻ�е��ר�Ŵ�����Ӧ��ָ����һ�ִ���������ֺͺ�����ȫ��ͬ������ֻҪ�ڴ���ϸ�������в��죬�Ϳ�����������۽ṹ��ͬ����������������кܴ�IJ��죬���²�����ʹ��Ҫ����ˣ��������Ʊ�ϸ�ڶ����ϸ��ܵġ�
3.6 ��ת�ƴ�
3.6.1 ����
����˫���ӷ�Ӧ��������������������Ӧ�����֮����뷢����ײ������������Ӳ������˿��ţ���ô��������һ�ַ��ӵ������ж����Ҳ���ܺ���һ�ַ��ӷ�����Ӧ�����磬���������軯����һ���������ڣ�Ҳ��������Ӧ��������Ϊ�軯����ȫ�������������Ե�ʡ������������л���ķ�Ӧ����ͳ�Ľ���취��ʹ�üȾ��������ԣ��־�����ˮ�Ե��ܼ������磬�״����Ҵ�����ͪ�����������ȡ�������Ҳ��һ�������ѣ�����������Щ�ܼ��е��ܽ�Ⱥ�С�����л����ֳ���������ˮ���������ַ����Ӽ����ܼ���������һ�����ܽ�ȣ�����ʹ��Ԫ���е�������רһ���ܼ������Ӷ�ʹ�����ӳ�Ϊ���Ե��������ӣ�������ȡ����Ӧ�����õ��ܼ�������ʹ�������ܼ�Ҳ��ȱ�㡣��Ҫ�Ǽ۸�����ھ��ƺ�����׳��ڱ�������ˮ״̬����ʱ����ˮ��Է�Ӧ�������š���Ӧ���ѻ��ա��ж��Ͳ������㡣
Ϊ�ˣ���60���ĩ�ַ�չ��һ�֡���ת�ƴ����л��ϳ��·����������ŵ��ǣ���һ�����Բ������������ܼ������ҳ�����Ҫ����ˮ�������ڶ���������ת�ƴ�����PTC���Ĵ��ڣ�ʹ��Ҫ�μӷ�Ӧ�������Ӿ��нϸߵķ�Ӧ���ԣ��Ӷ����ͷ�Ӧ�¶ȡ����̷�Ӧʱ�䡢���չ��̡���߲�Ʒ�����ʺ����������������ϡ�����������ͨ���ԣ��ɹ㷺Ӧ�������Ԫ��Ӧ����ת�ƴ�����ȱ������ת�ƴ����۸�Ϲ�ֻ����ʹ����ת�ƴ���������������ʡ����Ʋ�Ʒ������ȡ�ýϺþ���Ч��ʱ���ž��й�ҵӦ�ü�ֵ��������ˣ����ڹ�ҵ����ȡ�������м�ֵ�ijɹ���
3.6.2 ��ת�ƴ���
��ת�ƴ�������Ҫ������������������Ҫ��һ�����ܽ�����Ҫ�����Ӵ�ˮ������ת�Ƶ��л��ࣻ��һ����Ҫ�����ڸ����ӵ�Ѹ�ٷ�Ӧ��
��Ȼ��һ�־��й�ҵʹ�ü�ֵ����ת�ƴ���������߱�����������
��1�� �����٣�Ч�ʸߣ��������ᷢ��������ķ�Ӧ�����ĵ��������ڹ�����ʧȥת���ض����ӵ�������
��2�� �Ʊ���̫���ѣ��۸������
��3�� ����С�������ڶ��ַ�Ӧ��
3.6.3 ��ת�ƴ���Ӧ��
������ת�ƴ�ԭ�����Կ���������������ת�ƴ����γɿ������л�������ӶԵĶ������ͻ�������ɲ�����ת�ƴ������з�Ӧ�����������������Ԫ��Ӧ��ʵ���ܶࡣ
3.6.5.1 ��±�����IJ�����Ӧ��
���ȿ���(��CCl2)��������̼ϩ������Ǽ�������̼ԭ����Χֻ���������ӣ���һ���dz����õ�ȱ�����Լ������������ּӳɷ�Ӧ�������ȿ�������ˮ�⣬��ˮ�е������ڲ���1�롣�������ȿ����Ĵ�ͳ����Ҫ�������ˮ�������ܲ������������������ת�ƴ����Ĵ����£����������������������Ũ��Һ�����ö������ȶ��Ķ��ȿ������䷴Ӧ���̴������¡�
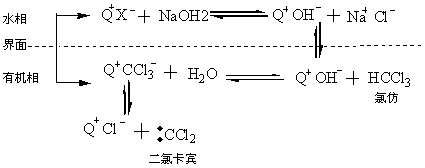
���ȷ����ɶ��ȿ����ķ�Ӧ����
����ˮ���м����Q+X����NaOH���ã����ɼ�識����Ӷ�Q+OH����������ȡ���л��࣬���ȷ����ö����ɶ��ȿ��������л����ж��ȿ���ˮ���������Ϊ�л����ж��ȿ��������ȼ�����δ���һ��ƽ����ϵ�У�������ȿ�����������һ����Ӧ�������л��������ܱ���ԭ�л��Դ�����֮�á����л����д�����ϩ����������̼���������ӡ�ȩ�������������Լ�ʱ���Ϳ��Է����ӳɷ�Ӧ�����ɶ������͵Ļ����
3.6.5.2 O-����������ĺϳɣ�
���������������ɶ������ȱ����������Ƶ��Ҵ���Һ�����ö��Ƶõġ��䷴Ӧʽ���£�
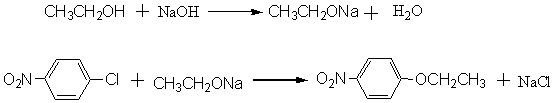 ���Ϲ��ղ�����ת�ƴ�����O-��������Ӧ������������Ӧ��Ҫ��ѹ�ȸ��м��ȼ�ʮСʱ����-�����ȱ���ת����ֻ��75%��Ҫ�ü�ѹ������δ��Ӧ�Ķ������ȱ����ܺĴ����⣬����ˮ�⸱��Ӧ�����ɶ�-�������ƣ���Һ�ࡣ�����ĵĶ�-�����ȱ��ƣ���-���������ѵ�����ֻ��85~88%��
���Ϲ��ղ�����ת�ƴ�����O-��������Ӧ������������Ӧ��Ҫ��ѹ�ȸ��м��ȼ�ʮСʱ����-�����ȱ���ת����ֻ��75%��Ҫ�ü�ѹ������δ��Ӧ�Ķ������ȱ����ܺĴ����⣬����ˮ�⸱��Ӧ�����ɶ�-�������ƣ���Һ�ࡣ�����ĵĶ�-�����ȱ��ƣ���-���������ѵ�����ֻ��85~88%��
������ת�ƴ������ڳ��¡���ѹ��ֻ�輸��Сʱ����-�����ȱ���ת���ʼ��ﵽ99%���ϣ���-���������ѵ����ʿɴ�92~94%�����ȴ�99%���ϡ���Ȼ��������Ϊ��ת�ƴ��������Q+X-��ԭ�������ڶ�-�����ȱ����Ҵ���ת��Ϊ�����ڶ�-�����ȱ��Ͷ�-���������ѵ�Q+C2H5O-���ӶԵ�Ե�ʡ�
3.6.5.3 O-��������Ӧ������ĺϳɣ�
����Ӷ�������������������-�����������ڼױ�-��������ˮ�������Ʊ��һ������ף��л���ɱ�����ʱ��
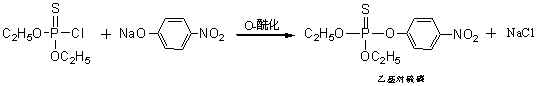
�����������ת�ƴ�������Ӧ�ٶȺ�����������ˮ�⸱��Ӧ�����ǣ�ֻҪ��������������װ�����Σ���25~40�淴Ӧ1Сʱ�����������ʿɴ�95~99.5%����Ȼ�������^���Q+X-�������ǽ������ڼױ��Ķ������������Ӷ�ת��������ڼױ���Q+-OC6H4NO2���ӶԵ�Ե�ʡ�
3.6.5.4 �����ӵ���ȡ��
���磬�Ӷ����������軯�������Ʊ��������裨ũҩ�м��壩ʱ���ڲ�����ת�ƴ��������Բ��÷����Ӽ����л��ܼ������ҿ����̷�Ӧʱ�䣬���ʿɴ�94~96%��
 3.6.6 Һ���̡�Һ������ת�ƴ�
3.6.6 Һ���̡�Һ������ת�ƴ�
���ǵ���ת�ƴ����۸���ѻ��գ��ַ�չ�˹�����ת�ƴ��������ǽ�����Ρ����l�Ρ����ѻ����������ӵ��ۺ����϶��õ��IJ�����ˮ��һ���л��ܼ��Ĺ�̬��ת�ƴ�������ת�ƴ���Ӧ��ˮ�ࡢ����������л���������֮����С�������������ֽ���������������������ŵ��ǣ�������㡢��Ӧ�������룬�����ɶ������ա����⣬���ַ���������ú���Դ���ܵͣ����������Զ�������������60������ִ����ѳɹ������ںϳ��贼�����һ����Ͱ�Ϣ�����ϵȷ�Ӧ��������ҵ��ļ�����Ȥ�����⣬���ִ����������ڰ����������칹��ķ��룬�����Թ��Ѿۺ�����������ڲ��Գƺϳɡ�
3.7 ������ϴ�
������ϴ�ָ�����ÿ����Թ��ɽ����������Ϊ��������Һ����л���Ӧ���о�����ķ��������ַ����ڹ�ҵ������ҪӦ�á�1977�꣬�������þ�����ϴ���Լ������900����л���ѧƷ������̽�����Լ24����Ҫ��ҵ���̡�
3.7.1 ���ɽ�����ѧ
3.7.1.1 ���ɽ������ص�
��õĹ��ɽ�����Ҫ��ͭ�����Ti����V����Cr����Mn����Fe����Co����Ni��ͭCu���������Mo����Ru���Rh����Pd����Ag���������W��ҿIr����Pt�ȡ����͵Ĺ��ɽ���ԭ�Ӷ������ڼ�����״�Ϻ��������������ڳɼ���1��s�����3��p�����5��d���������������£���9��������Ժ�9����λ��ɼ������磬蝹������ReH7[P(C2H6)2(C6H5)]2��������7��Re-H���ۼ�������Re-P��λ����������ԭ��һ����9����λ�������
3.7.1.2 18���ӹ���
������ɽ���ԭ�ӵ�9�����ܳɼ��Ĺ�����dz����ģ���������ϵ��ܵ�������18����������������DZ��͵ģ��ȶ��ģ������������������λ����ϡ���ʱ��λ���ȡ����ӦҪ������18����������Ͻ�������һ����������λ�壬����һ��16���ӵġ���λ�����͡��������������������ٺ�������λ���ϣ������ɱ��͵�18�������������18���ӹ���Ȼ���ھ�����ϴ���Ӧ�У�����������Ҫ����18��������
3.7.1.3 ���
������λ������ɽ���ԭ�ӵijɼ���ʽ������������һ��ȷ�����£�
��������λ�壺���ṩһ����������ɽ���ԭ���γɹ��ۼ������磬����������һ����������Ȼ��ȡ�
��������λ�壺���ṩ������������ɽ����γ���λ�������磬һ����̼������̼ԭ�ӣ�����ϩ��������˫�������������ӣ������ࣨ���е�ԭ�ӣ�����ࣨ������ԭ�ӣ������������̼ԭ�ӣ��ȵ��ӶԸ��壬�Լ�һ�������ӵȡ�
��������λ�壺���磬��-1-��ϩ����������һ�������Ӻ�����̼ԭ���ϵ�һ�����ӣ���
�ĵ�����λ�壺���磬��ϩ��������˫���ϵ��ĸ������ӣ���
3.7.2 ������ϴ���
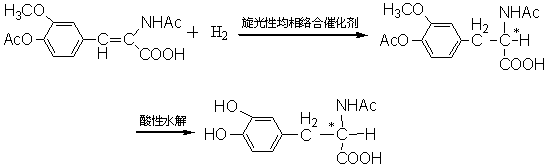
������ϴ��ܹ����ڶ��ֲ�ͬ���ͷ�Ӧ�İ��ؾ�����������ɽ���ԭ�����Բ�ͬ�ļ�̬���֣�����������ֲ�ͬ����λ���Թ��ۼ�����λ�����ֲ�ͬ�ļ������϶��������ֶ������ܲ�ͬ�Ĵ�������������Ƿ���̬�ģ���������ַ�Ӧ����ӷ����ض���һϵ�л�����Ӧ����ͨ����ѭ�����õ�Ŀ�IJ�����������ɴ��������ڲ�ͬ�ķ�Ӧ����������Ӳ�����Ҫ�ض��Ĺ��ɽ���ԭ�ӣ����һ���Ҫ����ض�����λ�壬����ʹ�������и�Ч�ʺ���ѡ���ԡ�����ϩ���ļ��⡢�ӳɡ���ۣ��Լ�һ����̼��̼���ϳɵȷ�Ӧ�����õĴ���������һ��Ҫ�����Ļ�ɼ�������λ�����ȶ����ɽ���ԭ�ӵĵͼ�����������λ����Ҫ�У�һ�Ȼ�̼�����ࡢ��࣬�ϴ��±�������Ӻ�CN�������ӵȡ�������λ�峣����ͨ����-���������-�����������������ԭ�ӽ�ϵġ����磬�������õĴ�����Ҫ�У��Ȼ�������좣����ڲ��ԳƼ��⣬����Ҫʹ�ò��ԳƵ����λ�塣�磺
3.7.3 ������ϴ��Ļ�����Ӧ
�ھ�����ϴ��ķ�Ӧ�������������ĵ�Ԫ��Ӧ��������λ��ѧ�ͽ����л���ѧ����һЩ������Ӧ������Щ������Ӧ�ʵ���ϣ���ɴ�ѭ�����͵õ�Ŀ�IJ�����������ɴ�������Ҫ�Ļ�����Ӧ�����¼��֡�
3.7.3.1 ��������
���ָ����һ����λ���ԼĹ��ۼ�����λ������ɽ���ԭ�ӽ�϶�������λ�����ķ�Ӧ�����Ǿ�����ϴ��в���ȱ�ٵķ�Ӧ�����磬�Ժ���������λ����⻯��Ϊ��������M-H��ʾ��������ϩ������Ƹ�̼��-ϩ��ʱ�����һ��������Ӧ������ϩ����ԭ�ӵ���λ��ϡ�
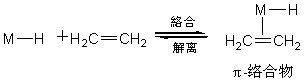
��������ϵ��淴Ӧ��������-��λ��֮��Ĺ��ۼ�����λ�巢������ʹ����λ���������н��������ķ�Ӧ�����Ǿ�����ϴ��о��������ķ�Ӧ��
3.7.3.2 ���������
����ָ��������ɽ���ԭ����λ��ϵ�˫��������ϩ������ϩ����Ȳ����������һ����̼����λ���е�˫�����е���-���������뵽��һ������-��λ���֮�䡣ָ��۵ĵڶ���������Ӧ������ϩ�IJ��뵽M-H��֮�䡣���磬������ϩ��۵ĵڶ���������Ӧ������ϩ�IJ��뵽M-H��֮��
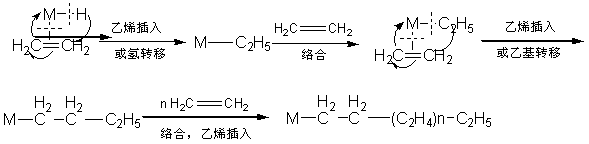 ���뷴ӦҲ���Կ�����һ����λ�壨������������λ��������λ�壩�ӹ��ɽ���ԭ����ת�Ƶ�һ������˫������λ�壨��������CH2��CH2������-λ�ϡ���˲��뷴ӦҲ������λ��ת�Ʒ�Ӧ�����ŷ�Ӧ��
���뷴ӦҲ���Կ�����һ����λ�壨������������λ��������λ�壩�ӹ��ɽ���ԭ����ת�Ƶ�һ������˫������λ�壨��������CH2��CH2������-λ�ϡ���˲��뷴ӦҲ������λ��ת�Ʒ�Ӧ�����ŷ�Ӧ��
����һ��ָ����һ����λ���ϵ���-H�����������ţ�ת�Ƶ����ɽ���ԭ�ӵĿ���λ�ϣ�ͬʱ����λ��-����֮��ļ����ѣ�ʹ����λ���Ϊ����˫���Ļ�����ӽ���������������
3.7.4 ������ϴ�����ȱ��
3.7.4.1 ������ϴ����ŵ�
��1������ѡ���Ժ� ��Ϊ������������Է���״̬���ڵġ�ÿһ���������Ӷ��Ǿ���ͬ�����ʵĻ��Ե�λ������һ�㶼�ǰ�����ṹ��ͻ��һ����ǿ����λ���á����⣬����̬�����ijߴ��С�����ڶ�����ŵ��л���Ӧ����ӣ���ͬһ˲��ֻ����һ�����������������ſ����������ӣ������������ڷ�Ӧ��λ�á�����ڷ�Ӧ������ѡ�����ṩ����������������������ͬ�����ı����ǷǾ�һ�ģ����ж��ֲ�ͬ�Ļ������ģ�����ͬʱ�������ֲ�ͬ����ķ�Ӧ�����粬�����ʹ��ѻ���
��2�������ĸ��� ���ھ�����ϴ������������Ĺ�������ԭ�Ӻ���λ��ľ���ɸѡ����ÿ���������Ӳ������кܸߵ�ѡ���ԣ������кܸߵĻ��ԡ���ˣ���Һ����ϴ�����Ũ��ԶԶ���ڹ���������������ֵ�Ũ�ȡ�
��3������ϵ��Ԥ���� ������ϴ����ڽṹ�Ϸ�Ϊ���Ĺ��ɽ���ԭ�Ӻ���λ�������֣����о�����ƴ���ϵʱ���Ͱ��ոı����Ľ���ԭ�Ӻı���λ���˼·�����������ܡ����������Ӵ���������������ɸѡ�нϺõ�Ԥ���ԡ�
���磬�ڱ�ϩ���������������ȩʱ�������H-Co(CO)3����������ӦҪ��140~180���25~30MPa�½��У�������/�춡ȩ�ı���ֻ��3~4�U1��������������������ʹ�������Ӧ���ڽ��º͵������½��У�����90~120���0.7~2.5MPa��������/�춡ȩ�ȿ���ߵ�10�U1�������������ԼΪ��ϩ��0.1%���ѽ������13��6��ֶ�ȩ��7��ֱ�ȩ�Ĺ������ɴ������Կ���������ĸ�ѡ���Ժ��ԡ�
3.7.4.2 �����ϴ���ȱ��
��1�������������⣬��ʹ�ù��������ʱ�ر���Ҫ��
��2�������������ϴ�����250�������Dz��ȶ��ģ���˷�Ӧ�¶Ȳ��˹��ߡ�
��3��������ϴ�һ���������Խ����н��еģ�����Ҫ��ʹ�����ֵ���ʴ���ϡ�
��4�������෴Ӧ���ر�����һ����̼Ϊ��ʼԭ�ϵ��ʻ��ϳɷ�Ӧ��������Ҫ�ߴ�30MPa�IJ���ѹ����
������ļ۸���ܹ�ǧ�������Ʊ�����ȩʱ�����ʧ�����ȩ�İ����֮һ����������ķ���Ҳ�����ܴ����ߺü�������������˾�����ϴ��Ĺ㷺Ӧ�á���ˣ�����Ӧ�ñ�������Ӵ���С�öֻ࣬ռȫ������Ӧ��20%���ҡ��������Ʒ��ȩ�ķе�Ƚϵͣ����������ӷ�ӦҺ������������Ӷ�����˴����Ļ��գ�ʹ�����������ڹ�ҵ����
3.8 ���л��ϳ�
3.8.1 ���ӵ������벨���Ĺ�ϵ
����һ�ֵ�Ų������������ԺͲ�����˫�����ʡ�����˵����������������ɡ�������С��Ԫ�ǹ��ӡ�һ�����������е�����e��ù����˶�ʱ�������IJ������������¹�ϵ��e=hc/����
3.8.2 ������ʵ�����
�������չ��ܵĹ��̽�����������������ʱ���Ӵ�������������ġ���̬��ԾǨ�������ϸߵļ���̬������ԾǨ�������з��ӵ�ת�������ӵ��ͷ����е��ӵ�ԾǨ������ת������Ҫ��������С��ֻ��ҪԶ�������ĵ��ܷ��伴�ɷ��������ӵ�������Ҫ��������ת������������Ҫ�������ķ��䡣�����еĵ��Ӵ�һ�����ӹ��ԾǨ����һ���ϸ��ܼ������ӹ�������������ܼ�����������̬����M�ͼ���̬����M�~����Ȼ������ͬ�ķ�����ɺͽṹ���������ǵĵ�������ȴ��ͬ�����˵���ԾǨ������̬�����㹻�ߵ��������п��ܷ�����ѧ��Ӧ������ԾǨ����Ҫ��������������10�����ϣ���Ҫ��λ�ڿɼ������������Ľϸ������Ĺ���䡣��ˣ��ɼ����������������⻯ѧ��Ӧ�������磬�ȷ��ӵĹ��������250kJ/mol(59.7kcal/mol)����˿ɼ����ֻ��Ҫ������С��479nm���Ϲ�������Ϳ���ʹ�ȷ��ӽ���Ϊ��ԭ�ӡ����磬һ��C-C ���Ľ�����ԼΪ335~377kJ(80~90kcal)����C-H���Ľ�����ԼΪ419kJ(100kcal)���������IJ���Ϊ320nmʱ�������е�����ԼΪ373kJ(89kcal)������������⣬ֻ��ʹC-C�����ѣ�������ʹC-H�����ѡ�Ӧ��˵�������DZ��������յĹ��Ӷ�������ѧ��Ӧ����Ϊ����̬���ӿ���ͨ������;��ʧȥ�������ָ�Ϊ��̬��
3.8.3 ����������
�⻯ѧ���������������ɡ�һ��������ֻ�б����������˵Ĺ������ܼ����⻯ѧ��Ӧ����һ��������һ������ֻ����һ�����ӡ�һ�����Ӿ�������ͨ������һ�����Ӷ���������̬���ӣ�Ȼ����ݲ�ͬ�����������ͬ�Ĺ��̡����磺
��1���������̵�ʱ�䣨Լ10-7~10-8s���������յĹ������·������������ө����⣬������ԾǨ��
��2��������������ײ���ѹ���ת��Ϊ���ӵ�ƽ���ܣ���ת��Ϊ���ܣ�����ʹ�������ӻ��
��3�������⻯ѧ��Ӧ��
���������ʣ������DZ�ʾ�⻯ѧ����Ч�ʵ����ȣ������¹�ʽ��ʾ��
��=��λ�����λʱ���ڲμӷ�Ӧ�ķ�����/��λ�����λʱ�������յĹ�����
һ���⻯ѧ��Ӧ���������ʴ�С���뷴Ӧ�ĵĽṹ���ʺͷ�Ӧ�����������йء�������������⻯ѧ��Ӧ���������������0~1֮�䡣�������෴Ӧ�����ĵĹ���̫�࣬���ҷ�Ӧ�ٶ�̫������ҵ�Ϻ��ٲ��á�
���ǣ�����������Ӧ������������ʿ��Ըߴ�10�ļ��η������磬�ȷ��Ӻ�������ڿɼ���������£������Ȼ���Ĺ��������ʿɸߴ�109���ҡ��䷴Ӧ�������£�
������Ϊһ���ȷ�������һ�����Ӻ����������������һ���������������ӷ�Ӧ�����Ȼ����һ�����������������������ȷ��ӷ�Ӧ�������Ȼ����һ�����������������������ֹ�ĸ���Ӧ�����������������Ӧ�Կɼ���ѭ����ȥ�����ʹ��ÿ����һ�������ӿ������ɼ���������ϵ��Ȼ�����ӡ�
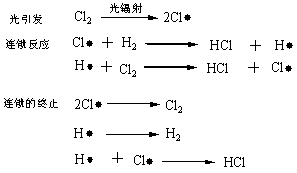
�ڹ�ҵ�ϣ����л��ϳ���Ҫ����������Ӧ����Ϊ���Ĺ��������ʸߣ����ĵ������١�Ӧ��ָ������Ʒ�Ĺ��������������������йأ������������������������йأ�����������ȫ��ͬ�ĸ��
3.8.4 �⻯ѧ��Ӧ����ҪӰ������
�⻯ѧ��Ӧ����ҪӰ��������һ���Ȼ�ѧ��Ӧ������ȫһ�����ֶ�Ҫ�������¡�
3.8.4.1 ������Դ
һ����Ȼ�ѧ��Ӧ�������������������ṩ��Ӧ��������Ҫ�Ļ�ܡ��Ȼ�ѧ��ӦҪ��Ӧ���������ܽ��͡��⻯ѧ��Ӧ��ͨ�����ӵ�����ʹ��Ӧ���ijһ���ż������ٽ���Ӧ�Ľ��С���Ӧ���������е��������Ը�����ʼ��Ӧ�������е�������
3.8.4.2 ��IJ�����Դ
�����ļ�����Ч�����Ǹ��ݱ������ļ�����Ҫ��������ȷ���ġ����磬�ȷ��ӵĹ��������250kJ/mol(59.7kcal/mol)������Ҫ����С��479nm���Ϲ������⣬��˿���ʹ�ø����������չ����Ϊ��Դ��������������(NOCl)��Һ����ΪNO����C1��ʱ����Ҫ����⣬��ʱ����ʹ�ø�ѹˮ������Ϊ��Դ����Ϊ�������ܷ���253.7nm������⡣����ӵĹ��������234kJ/mol(55.9kcal/mol)��ֻ��Ҫ����С512nm�Ŀɼ���(��-����)���ɡ�
3.8.4.3 ����ǿ��
�⻯ѧ��Ӧ���ٶ���Ҫȡ���ڹ�ķ���ǿ�ȡ���Щ�Ĺ⻯ѧ��Ӧ�����ٶ�ֻȡ���ڹ�ķ���ǿ�ȣ����뷴Ӧ���Ũ���ء�
3.8.4.4 �¶�
����һ���л���Ӧ���¶�ÿ����10�淴Ӧ�ٶ�Լ����2-3������������⻯ѧ������Ӧ���ٶȣ������¶ȵ�Ӱ���С�����л��ϳɷ�Ӧ�У����Ӱѷ��ӻ�������Ż��м����ǹ⻯ѧ��Ӧ����ʱ�������������Ӧ�ٶȵ��Ǻ���ķǹ⻯ѧ���裬��ô�¶ȵ�Ӱ�콫��һ���Ȼ�ѧ��Ӧ���ơ�
3.8.4.5 �ܼ�
�ܼ��Թ⻯ѧ��Ӧ��Ӱ���о��û��ܲ���֡��������л��ϳ�����������������Ӧ��˵������ѡ��ᵼ����������ٵ��ܼ�����Ӧѡ�������ڱ�����������ܼ������磬�ڼױ���������ʱ������CCl4���ܼ����ⲻ������Ϊ�ڷǼ����ܼ����ȷ��ӽ����Ϊ��ԭ�ӣ�����ΪCCl4��ͨ�����½�����Ӧ���ױ�����ԭ�ӣ��Ӷ������˹�����Ч�ʡ�
3.9 ����л��ϳ�
3.9.1 ����
70������淢�˹������⣬�ƶ����й����������ķ��档���⣬ʯ�ͼ۸����ǣ�������Դ��ҵԭ�ϼ۸�Ҳ��֮���ǡ������ʹ��ҵ�����м����������ۡ�Ҫ���������뿼��������ʡ��Դ��ʡ��Դ���ڴ˱����£�����л��ϳɣ�����������Ч�ʸߣ��ܹ����ڶ�����Դ����������������ص���������ӡ��˺���л��ϳ��ڼ����Ϻ������϶����˺ܴ�չ���ݳ���ͳ�ƣ���80�����������л��ϳɼ����Ѿ���ҵ��������ʮ������Ѿ���ɺ��������ԵĻ���ʮ����������л��ϳɿ����õķ�Ӧ���͡�ͬһ���л�ԭ�ϣ��ڲ��������¿����ɶ��ֲ�ͬ������磬�������ڲ�ͬ�����½���������ԭ�ɷֱ��Ƶñ����������ӡ����������Ѻ��������ĸ���Ʒ�����⣬��Щ������Ʒ���õ���л��ϳɷ���ԭ���ã���һ��ֱ���Ƶò�Ʒ���ȷǵ���л��ϳɷ����ж��ص��ŵ㡣���磬�����ȵ�����������ȫ�����ᡢ�������ȡ������ѿ�Ӻ��һ���ѿ̪���Ѷ��ᵥ������������ż���ƹ����˫������������/��ԭ�ƶԱ����ӡ���ϩ��ļ���ż���Ƽ�����ȡ�������Щ��Ʒ���õ���л���ɷ�����Ϊ���ú��������磬��ϩ��ļ���ż���Ƽ����棬����Ӣ�ȹ��������10��ֵ�װ�ã�����ѽϹ���ձ�Ҳ�ѹ�ҵ����Ԥ�ƽ�����л��ϳɽ����и���ķ�չ��
3.9.2 �����̵Ļ�����Ӧ
���еĵ��۶�����������Һ��Ӵ��ĵ缫���绯ѧ�������ڵ缫����Һ�Ľ����Ϸ����ġ����������л���Ӧ��R-H����ʧ�������ã���������ת��Ϊ�����ӻ���R-H��+�����������л���Ӧ����õ������ã���ԭ���ã���ת��Ϊ�����ӻ�[R-H]-��

3.9.3 �����̵ķ�Ӧ˳��
��Ӧ˳��ָ����Ϊ�˵õ�Ŀ�IJ����ʼ��Ӧ���ڵ������������ĵ绯ѧ����(E)�ͻ�ѧ����(C)��˳����Ӧ���̡����磬��ϩ����������������ۣ�ż�������ɼ�����ķ�Ӧ˳����Ա�ʾ��ͼ��ʾ��
��ˣ��ɱ�ϩ�����ɼ�����������ĸ���Ӧ���̣��䷴Ӧ˳��ֱ�Ϊ��ECC��ECCC��ECECC��ECCEC������������Ӧ����������ECE��EEC��CEC�Ȳ�ͬ�ķ�Ӧ˳��
���������ɱ�ϩ�����ɼ�������ܷ�Ӧʽ�ɼ�ʾ���£�
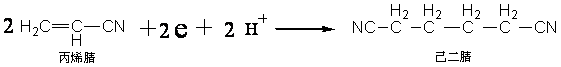
 ��ϩ����������(ż��)���ɼ�����ķ�Ӧ˳��
��ϩ����������(ż��)���ɼ�����ķ�Ӧ˳��
���������Կ�������ϩ��ĵ�������۵��л���Ӧ�������������ģ�����缫�����������缫������һ���缫�������������������������л���Ӧ��ֻ������������л���Ӧ�ṩ���ӣ�����缫�����������缫�������������л��ϳɹ���ֻ����һ���缫����Ŀ���л���Ӧ��ֻ���������̲����������缫���μ��л���Ӧ�����磬�������������ɶԱ�����Ȼ����������ԭ�ɶԱ����ӡ�
3.9.4 ��ⷴӦ��ȫ����
�������г��绯ѧ��Ӧ(E)�ͻ�ѧ��Ӧ(C)���⣬���漰�������������̣�������ɢ���������������ȡ������Ա�ϩ�水ECC˳�����ɼ�����Ϊ������ȫ�������ٰ��������߸����裬��ͼ��ʾ��
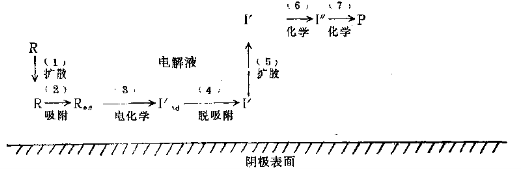
�ɱ�ϩ�水ECC���ɼ������ȫ����
��1����Ӧ�����R������ϩ�棩�ڵ��Һ��������ɢ��Ӿ�������������档
��2��R�����������ϱ�������Ϊ������Ӧ��Rad����������Ҫ��������������ʱҲ�л�ѧ������
��3��Rad������֮�䷢������ת�Ʒ�Ӧ�����ɱ��������м���I`ad������ϩ��õ�������-CH2-CH2-CN�����ӻ�����
��4��I`ad��������������������Ϊ���������м���I`��
��5�����������I`����Һ����ɢ��Ӿ�����뿪�������档
��6��I`�ڵ��Һ�з�����ѧ��Ӧ�������м���I``
��7���м���l`�ڵ��Һ�н�һ��������ѧ��Ӧ�����ɲ���P����-CH2-CH2-CN�Ķ������ɼ����棩�����ˣ������ĵ�ⷴӦȫ������ɡ�
����(1)��(5)�������ƶ����������̡��ڹ�ҵ�����У����������Ϊ���Ʒ�Ӧ�ٶȵ���Ҫ���ء�����ϵ�����۵���ƺͲ���������ȷ����������Ϊ��ѧ�������������ǡ�
����(3)�ǵ绯ѧ���̣����ǵ�ⷴӦ�в���Ҫ�Ĺ��̣�Ҳ���������۵����ġ�
����(6)��(7)�ǻ�ѧ���̡������л���ѧ���о���������ȷ���ķ�Ӧ������Ӧ�ø��ű�Ҫ�ĵ绯ѧ���̺��������̡�
����(2)��(4)�����������������̣������л�������������������йصij������⣬һ�㲻��̫��Ŀ��ǡ�
3.9.6 ����л��ϳɵ���ȱ��
��Ҫ�ŵ�
��1�������ೡ�Ͼ���ѡ���Ժ�������(����)��
��2������Ҫʹ�ü۸�Ϲ���������ͻ�ԭ����
��3����Ӧ�����º͵������½��С�
��4���ǽ��ܵķ��������ѱ������������ͻ�ԭ�����õöࡣ
��5����������������෴Ӧ��
��Ҫȱ�㣺
��1������豸���ӣ�ר����ǿ��
��2��Ӱ�����ض࣬���������ѡ��͵绯ѧ���̼����Ĵ����Ƚϸ��ӡ�
��3��������Ҫʹ���л��ܼ���
��4�����ڿ��ÿ�����������������������ԭ���ķ�Ӧ���������
��5������dzɱ�����Ҫ���֡�����Ҫ��һ��ת���ʽӽ�100%�ķ�Ӧ������Ч�ʵ͡���Ѹߡ�
3.9.7 ����л��ϳɵĹ�ҵӦ��ʵ�����������������
����������������66����Ҫ�м��壬�ڹ�ҵ����Ҫ����������������
��1��������������������ˮ�����Ĵ����£���200~300����ˮ�����ɼ�������������ˮ�����Ѷ��档
��2������ϩ���м���l,4-���ȶ�ϩ��������軯��
��3������ϩ�ھ�����ϴ���������ֱ�����軯.
��4����ϩ��ĵ���������Ƽ����档
�����ַ���������ȱ�㣬���������ܺ��ϱ�ϩ�淨��ͣ���ĵ����ֻռ�����ѵ�5%����ˣ����ǵ�ѽϸߵ��ձ�Ҳ�Ѳ��ô˷���
��ϩ�淨��������ɽ����˾���ȿ����ġ��������õĵ����Ǹ�Ĥʽ���ۡ���Ĥ���ɻǻ�����ϩ��֬���Ƴɵ����ӽ���Ĥ���缫����һ��������һ��������˫��ʽ�缫�⡣�����Dz���������Ǧ��������ʽ�ǰ��ѹ�˻��͡�ÿ��缫��������0.93m2��������С�������缫���Ĥ�þ۱�ϩ�Ƶķ�����������������ҵ��Һ������DZ�ϩ��40%�����һ�臨Լױ�������34%��ˮ26%�������ҵĵ��Һ��ϡ���ᡣ���ֵ��Һ�ֱ����������������Һ������ҡ��������Һ���������Ѷ��������Ϊ90~93%���۵����ܶ�50~100A/L������Ч��90~92%�����6.6~7.7kw/Kg��
�������ҵ����˾���Ա�ϩ�淨�����˲����µ�ר�������а����缫�͵��Һ�ĸĽ������ø�Ĥ�Լ����۵���Ƶȡ�