第24章 周环反应
一 周环反应
1.定义:在最近的五十年里,有机化学家研究有机化学机理,主要有两种。一种是游离基型反应,一种是离子型反应,它们都生成稳定或不稳定的中间体。
离子型或游离基型反应: 反应物→中间体→产物
另一种机理是,在反应中不形成离子或游离基的中间体,而认为是有电子重新组织经过四或六中心环的过渡态而进行的。这类反应不受溶剂极性的影响,不被碱或酸所催化,没有发现任何引发剂对反应有什么关系。这类反应似乎表明化学键的断裂和生成是同时发生的。这种一步完成的多种心反应叫周环反应。
周环反应:反应物→产物
2.周环反应的特征:
①反应进行的动力,是加热或光照。
②反应进行时,有两个以上的键同时断裂或形成,是多中心一步反应。
③反应时作用物的变化有突出的立体选择性。
④在反应过渡态中原子排列是高度有序的。
二. 分子轨道理论
几个原子轨道线性组合,形成几个分子轨道,比原子轨道能量低的为成键轨道,比原子轨道能量高的为反键轨道。其电子填充符合Pauli原理和Hund规则。
σ轨道:
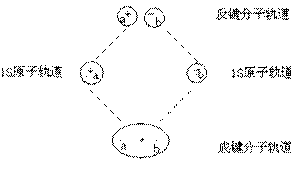
Л轨道:

丁二烯的分子轨道: 镜面 节面
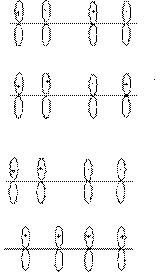
直链共轭多烯烃分子轨道特点:
①节面数:若共轭多烯烃有几个原子,它的n个轨道就有n-1个节面。
②轨道的节面越多,能量越高。
③高一能级的轨道和低一能级的轨道的对称性是相反的。
④图中的共轭多烯烃的对称性都是指类顺型(像顺型)的。
三.前线轨道

福井认为最高的已占分子轨道(HOMO)上的电子被束缚得最松弛,最容易激发到能量最低的空轨道(LUMO)中去。这些轨道是处于前线轨道(FMO),前线轨道理论认为:化学键的形成主要是由FMO的相互作用决定的。分子的HOMO与LUMO能量接近,容易组成新轨道。
§1.电环化反应
1.定义:在n个Л电子的线型共轭体系中,在其两端点之间生成一个单键的反应及其逆过程称为电环化反应。电环化反应中,多烯烃的一个Л键变成环烯烃里的一个σ键。
如: 
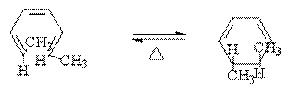
(Z,E)2,4-己二烯
根据微观可逆性原则,正反应和逆反应所经过的途径是相同的。
2. 4n个Л电子共轭烯烃的电环化反应
以(Z,E)2,4-己二烯为例.
①加热时: 
根据前线轨道理论,电环化反应取决于能量最高的电子占有轨道(HOMO)。(Z,E)2,4-己二烯4个 π 电子共轭,其HOMO为 Ψ2: 
Woodward和Hoffman指出:“当反应物与产物的轨道对称性相合(轨道对称性匹配或者说相位相同)时,反应易于发生,并不相合时,反应难于发生。”顺旋时反应物与产物对称性相合。前线轨道理论,只看HOMO的两头的相位,不管中间。

因此,对称性允许的,与实验一样。


对旋时反应物与产物的轨道对称性不相合,因此是对称性 禁阻的。
②光照时:“

光照时,电子激发HOMO为Ψ3: 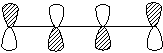
 对称性允许易反应
对称性允许易反应
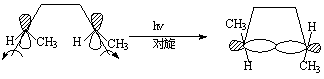 对称性允许易反应
对称性允许易反应
与事实一致


总结:4nЛ电子体系: 加热 顺旋
光照 对旋
如:

3. 4n+2个π电子线型共轭烃的电环化
以(2,2,E)2,4,6-辛三烯为例
基态参加反应
①加热时:HOMO:


②光照时:

HOMO:


总结:4n+2电子体系 加热 对旋 允许
光照 顺旋 允许

4.影响电环化的因素
①空间位阻
空间位阻大的不易形成

②热力学稳定性
电环化反应是可逆反应,反应向哪一方向进行取决于共轭多烯烃和环烯烃的热力学稳定性。环丁烯由于环的张力较大,与1,3-丁二烯相比是不稳定的体系,因此,在热反应中通常只观察到环丁烯的开环。
丁二烯关环成环丁烯的反应,只知道很少几个例子:Z,E-1,3-环辛二烯由于含有环内反式双键,张力很大,加热到80度可以环化:

但在光的作用下,丁二烯及其衍生物按对旋的方式变为环丁烯及其衍生物。这一反应大多数是形成关环化和物,而逆向开环反应则不易发生。这是由于双烯比烯吸收光的力量更强,因此双烯容易被激化。由于逆向反应是热禁阻的,所以应当预料到,产物非常稳定。这是合成环系的有效方法。


§2. 环加成
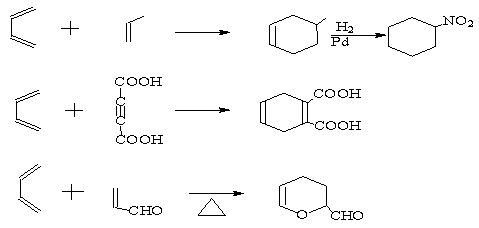
两分子烯烃或多烯烃变成环状化合物的反应叫做环加成。
例如:

环加成可以根据反应物中的π电子的数目分类。两分子烯烃变成环丁烷的反应叫做[2+2]环化加成,一分子丁二烯与一分子乙烯变成环己烯的反应叫做[4+2]环化加成。 反应就是[4+2]环化加成反应。
一.[2+2]环化加成反应
两分子乙烯变成环丁烷时,两个π键变成两个σ键。成键要求两个轨道重叠。一个轨道只能容纳两个电子,因此,一个乙烯分子的已占据轨道只能与另一个乙烯分子的未占轨道重叠。
假定两个乙烯分子面对面相互接近。在加热反应中最高占有轨道为π轨道,另一个乙烯分子的最低未占轨道为  轨道,它们的相位不同,因此是轨道对称性禁阻的。光反应是两个激发态分子的各自HOMO轨道作用决定的(自已的观点)
轨道,它们的相位不同,因此是轨道对称性禁阻的。光反应是两个激发态分子的各自HOMO轨道作用决定的(自已的观点)

在光反应中一个处于激发态的乙烯分子的最高已占轨道为 轨道,另一个处于基态的乙烯分子的最低未占轨道也是 轨道,它们的位相相同,可以重叠成键。因此,是轨道对称性允许的。
光化反应是一个处于激发态的分子与另一个处于基态(既然是光反应,则两分子都应为激发态,一个处于基态,一个处于激发态, 这种情况不合乎逻辑。 自己的观点)的分子之间的反应(尚龙海)。其它烯烃的轨道对称性与乙烯相同,因此,[2+2]环化加成在面对面的情况下,热反应是禁阻的,光反应是允许的。

实验事实与理论推测完全符合。例如,(2)-丁烯-2在光照下生成1,2,3,4-四个环丁烷的两种异构体。

二.[4+2]环化加成
最简单的[4+2]环化加成是1,3-丁二烯与乙烯加成反应,这是一个热反应,假定丁二烯分子与乙烯分子面对面互相接近,丁二烯的最高已占轨道π2与乙烯的最低未占轨道或丁二烯的最低未占轨道与乙烯的最高已占轨道都可以重叠成键,因此,[4+2]环加成对于热反应是轨道对称性允许的。

[4+2]环化加成反应(热反应)允许
[4+2]环化加成反应具有下列特点:
(i)是可逆的反应。利用逆反应有时可以得到一些用别的方法难以合成的化合物。例如:


(ii)加成时是立体专一性的,无例外的都是顺式加成。

(iii)如亲双烯分子上还有其它的不饱和基团时,加成后不饱和基团是靠近于新产生的双键一面。它是一个经验规则,称为阿尔德规则,也称不饱和性最大积累规则。[夏炽中译[美]F.A.凯里等“高等有机化学“A卷P367]。例如,两个分子的环戊二烯合成反应时,一个分子供给一个双键,作为亲双烯使用。加成时生成二环戊二烯, 理论上应有两种取向: 一种是外型(Oxo)的, 即原有的双键和新生成的双键处在离的较远的位置; 另一种是双键处在离的较近的位置, 这叫作内型(Endo)的,双烯合成主要产生内型产物。(邢其毅主编”基础有机化学示范教学“P308,胡宏纹讲的”周环反应“解释为次基轨道的结果)。
在形成内型产物的过程中,一个环戊二烯的HOMO和另一个环戊二烯的LUMO除了直接成键原子上的P轨道重叠外,还有在不直接成键原子上的P轨道之间的次级相互作用,这种次级相互作用的对称性是匹配的。这样相互作用的结果同样可以使过渡态的能量降低,因而有利于这种内型异物的生成。

这一反应也是可逆的。事实上,环戊二烯是不稳定,在实验室内,是用二聚体保存,等到使用时,加热使之解聚,发生逆向的双烯合成(邢其毅“基础有机化学“P829)。
(iv)区域择向性
假如双烯与亲双烯都是不对称的,加成就有一个位置选择性问题。一般地说,“邻”和“对”的定向较为有利,这种取向用定性的分子轨道理论理解为最有利的过渡态,应在双烯的HOMO和亲双烯的LUMO之间有最强烈的相互作用。[K.N.Houk,J.Am.chem.soc.95,4092(1993)]在X是推电子基团和Y是拉电子基团时(这是常见的情况),亲双烯体的LUMO轨道将与双烯的HOMO强烈地作用。这些分子轨道的系数在双烯的4位碳和亲双烯的2位碳最大。这样,键合给“邻”异构体是有利的,因为双烯体的C-4和亲双烯体的C-2之间成键构成了这一异构体。(王积涛译[美]F.A凯里等“高等有机化学”B卷P162-163)

(v)凡是双键或三键上有吸电子取代基(  ) 的化合物都容易与二烯起加成反应。
) 的化合物都容易与二烯起加成反应。
吸电子基团,可以降低HOMO和LUMO的能量;给电子基团可以增高HOMO和LUMO
的能量;共轭碳-碳链增长可以增高HOMO的能量,降低LUMO的能量。
有机合成中的应用:
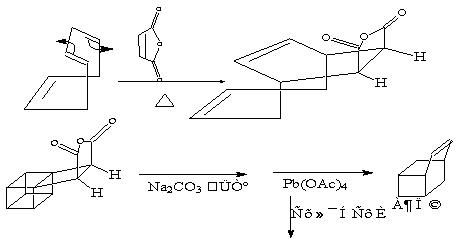
三.1,3-偶极加成
重氮甲烷的结构为: 
通过电子的转移可以具有1,3-偶极结构(即原子1和3上分别带负电荷和正电荷。


碳原子上的P电子与氮氮双键上的π电子共轭,这是一个有三个原子的四个电子所组成的共轭体系,与二烯烃相似,它也可以参加[4+2]环化加成反应。例如:

臭氧的电子结构为:框里圆点表示电子,框外圆点表示孤对电子:
臭氧化反应也是一种1,3-偶极加成:

三. 其它


总结:(K1+K2)π电子体系: 4n个π电子 光照 允许
4n+2个π电子 加热 允许
§3.σ迁移反应
一. 定义、命名:
1.定义:1,3己二烯在加热时变成2,4-己二烯,C1上的一个氢原子迁移到C5上,σ-键也随着移动:

碳碳之间的σ-键也可以迁移,例如:

这些反应都是协同反应,旧的σ键断裂、新的σ-键生成和π-键的移动是同时进行的。反应在加热或光照下进行,不需要催化剂。
一端或两端与π-键体系相连接的σ-键经过环状过渡迁移到新的位置,π-键体系也随之
重新组合,这类反应统称σ-迁移。
2.命名:σ迁移的命名是以作用物中发生迁移的σ键作标准,从它的两端开始分别编号。把新生成的σ键所连接的两个原子的位置i,j放在方括号中叫做[i,j]迁移。如上面两个反应分别为[1.5]迁移和[3.3]迁移。
二.氢原子参加的[i,j]迁移
1.同面反应异面反应
①同面反应:迁移前后的σ键在共轭平面同一侧。

②异面反应迁移前后的σ键在共轭平面的异侧。

2.氢原子参加的[1,j ]迁移 j个碳原子共轭烯自由基的HOMO
假定C-H键断裂后生成一个氢原子和一个含j个碳原子的自由基(这是分析问题时的假定,并不代表反应历程),自由基的轨道为:
如:[1,3]迁移:烯丙基自由基轨道为:

戊二烯自由基轨道为:
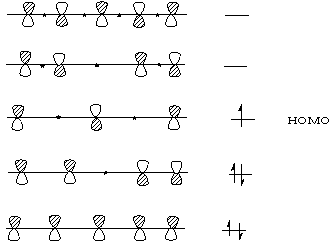
结论:

氢原子参加的[1,j]迁移j个碳的共轭自由基的HOMO
3.[1.3] σ键氢迁移
烯丙基自由基HOMO为:
同面禁阻 异面对称性允许

[1.3]σ键氢迁移,同面特征是禁阻的,异面转移是允许的。但由于分子的几何构型,氢从一边转移到平面的另一边所需的能量较高,异面迁移可能使对称性允许过程变的困难或不可能。
4. [1.5] σ键氢迁移
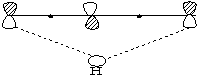
同面允许,异面禁阻。
如:1-氘茚在加热至200℃时,可得到2-氘茚,它是经过氘的σ键[1.5]迁移,而后又经过氢的σ键[1,5]迁移而得到的。

5. [1.7]σ迁移 同面禁阻,异面允许
三.碳原子参加的[i,j ]迁移
上述迁移的是C-Hσ键,与氢的迁移类似,反应是烷基的迁移,即C-Cσ键的迁移。氢的迁移时只有同面成键或异面成键问题,涉及到烷基碳的迁移时,既有面的问题还有迁移碳原子轨道以原有的一瓣成键(构型保持)还是以不成键的另一瓣去形成新键(构型反转)的问题。
1.[1.3]迁移
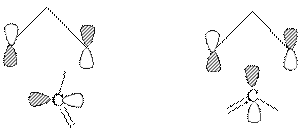
同面,构型反转(转化),允许 同面,构型保持,禁阻
如:
2.[1.5]迁移

同面,构型反转(转化),禁阻 同面,构型保持,允许
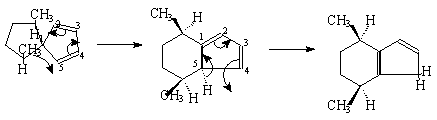
同面,构型保持,允许 同面,允许
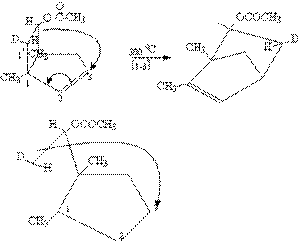
3.[3.3`]σ键迁移
最简单的[3.3`]σ键迁移为:

假定σ键破裂,生成两个烯丙自由基,其最高已占轨道中,3、3`两个碳原子上P轨道
最靠近的一瓣位相相同,可以重叠。在碳原子1和1`之间的键开始破裂时,3,3`之间就开始成键。

这样的过渡状态是轨道对称性允许的,空间条件也是可能的。[3,3]迁移是例证最多的σ键迁移反应。Cope重排和Claisen重排就是典型的例子。
①Cope重排:
3,4-二甲基-1,5-正二烯有两个手性碳原子经Cope重排主要可以得顺,反(Z,E)-2,6-辛烯

3,4-二甲基-1,5-己二烯 0.3%(E,E)式 99.7%(Z,E)式
这说明反应的过渡态为椅式:

②Claisen重排
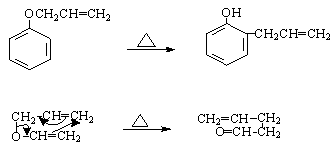
这是一种有碳氧键参加的[3,3]迁移反应。
苯酚的烯丙醚加热时,烯丙基迁移到邻位碳原子上,γ碳原子与苯环的邻位相连接。

如邻位全部被占具的情况下,全得对位产物。


а碳原子用同位素标记,然后进行重排,也得到同样的结论。
