��ʮ���� ��������������
��1����������
�����к�����������NO2���Ļ������Ϊ�������������ȡ�������ϵ������ɵķ���������������ArNO2����������Ӧ���ã�����Ҫ�Ĺ�ҵ��;������ȡ�����������е������ɵ�֬��������������RNO2��ֻ�м����Ļ������й�ҵ��;�⣬������;���١�
1.1 ����������Ľṹ
����������������нϸߵ�ż����,�����ⶨ������ԭ�Ӻ͵�ԭ��֮��ľ������,�Ӽۼ����۹۵㿴����ԭ�ӵ�sp2�ӻ�����γ�������ƽ���������δ�μ��ӻ���һ�Ե��ӵ�p�����ÿ����ԭ�ӵ�p����γɹ�����ϵ����ˣ�����������ķ��ӽṹ���Ա�ʾ���£�

����ṹʽ��
����һ������ɣ�ÿ��������½����ɣ���������������ߵ�ż��������ϵ������R�IJ�ͬ��ż������3.5D��4.0D֮�䣬���������������ż������,�������ͬ��������ͪ�е�ߣ��ӷ����������������飨MW61���е�101��,��ͪ��MW58���е�56��������أ���ˮ���ܽ�ȵͣ���ˮ����������ı�����Һ��������������10%������ͪ��ȫ����ˮ��
1.2 ������������������������ʣ���ѧ��
1.3 ����������������ķ�Ӧ
���������������������ܻ�ԭ�ɷ����������߿���ת��ɶ����м������������л��ϳ������൱����Ҫ�ԡ�����������������������ЧӦ���Է����ϵ���ȡ����Ӧ�л���á�
�� ��ԭ������������������λ�ԭΪ�������������N-����ȡ���ǰ�������

������������ǿ������£����������Ƚ�����ԭ������Ϊ�������������������ṩ���ӡ����磺

�����Ի���������Һ��N-�����ǰ���ԭ���ʺ���������ΪN-�����ǰ������磺

�ڼ�����Һ��������������N-�����ǰ�������ԭ�����ʶ���������ˣ�����Ϊ����ż������������ԭ���������磺

�� �����ϵ���ȡ����Ӧ
�����ϴ��������ӵ�ȡ����ʱ��ʹ���ϵĵ������ܶȽ��ͣ����Է�����ȡ����Ӧ����������ȡ����Ӧ��������ǿ�����ӻ��ţ���ʹ���������Խ�����ȡ����Ӧ��������������λ���λ�ϵ�±ԭ���ױ����Լ�ȡ������ȥ����Ҳ������±ԭ�ӣ����磺


��Ӧ�������ӳ�-��ȥ���� �������������Լ����ɼӳɲ��Ȼ����ȥ�����ڴ���һ�Ե����뿪����

��3��֬��������������
֬��������������Ϊ��ɫ��������Һ�壬��ˮ�е��ܽ�Ⱥ�С�������ڶ����л��ܼ������������������һ�����õ��ܼ���
��������������Ц�-H�IJ�����������������ˮ��Һ���뵪���γɶ�̬ƽ�⣺������Ľṹ���������ƣ�

�� ����(Henry)��Ӧ:

������������ȡ���������-H�����Ե����ԣ��乲���Ľṹ���ù���ʽ��ʾ��


�� ��ԭ��

�� �����������
��±������

���ߵı�����±�����Ľṹ�йأ�����������������Ϊ��
��±��������±��������±����
±������±�ر�ȡ���������ǣ�I>Br>Cl>F
��2 �ص���ż��������
1.���ص�����
1.1. �ṹ�����ķ���ʽ��CH2N2���ص�����Ľṹ�Ƚ��ر𣬸��������������������� һ�����ͷ��ӣ�����û��һ���ṹ�ܱȽ������ı�ʾ���Ľṹ���������������м���ʽ����ʾ��


ʵ����������������������ż���ز�̫�ߣ����������Ϊ�ص�����ķ�����һ��̼ԭ�Ӻ�������ԭ���ϵ�p���ӻ����ص����γ�����ԭ�ӵ���������������˼���ƽ������
1.2 �ص�������Ʊ���
�ص���������üװ���������ֱ�������Ƶã���ö��ַdz�������Ʊ���


1.3 �ص���������ʺͷ�Ӧ
�ص�������һ���ж������壬���б�ը�ԣ��������Ʊ���ʹ��ʱ��Ҫ�ر�ע�ⰲȫ�������������ѣ����ұȽ��ȶ���һ���ʹ������������Һ���ص�����dz����ã��ܹ������������͵ķ�Ӧ�����л��ϳ�����һ����Ҫ��������ļ��������������л��ܼ�����Ӧ�ٶȿ첻��Ҫ�������ֽ��N2,�������⣬���ʺܸߣ��䷴Ӧ���£�
1.�˷�Ӧ��
�������Ի�����ķ�Ӧ��




�����������ã�










Wolff(�ֶ���)�ص�ͪ����Ϊϩͪ��

����������˹��(Arndt-Eistert)��Ӧ��

�����Ӧ��Arndt-Eistert��Ӧ�����а����ص�ͪ��wolff���ţ��÷�Ӧ�ǽ����������ĸ�һ��ͬϵ���̼������������Ҫ����֮һ��

����ȩͪ��Ӧ
�ص�������ȩͪ��Ӧ�ܵõ���ԭȩͪ��һ��̼ԭ�ӵ�ͪ��




��4�� 1��3��ż�����ӳɷ�Ӧ
�ص��������1��3��ż����͵�����C=C�����ѷ�����Ӧ��ֻ�й�����ϵ�е�C=C�����籽��ϩ������-̼ԭ�����������ӻ��ŵ�˫�����������¡������͵��ʻ�����������Ǻ��ص�������ӳ����ɶ������������


��5���γɿ�����Carbene :CH2��

������һ����Ӧ�м��������ԣ�ֻ���ڷ�Ӧ�ж��ݵĴ��ڣ�Լ�ܴ���һ���ӣ������Ľṹ��һ��̼��������������ɡ�̼���ж����µ��ӣ�����������µ��ӳɶԣ������෴ 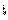 ���Ƶ���̬������singlet Carbene������������µ��Ӳ��ɶԣ�����ƽ�У�������̬������triplet Carbene��
���Ƶ���̬������singlet Carbene������������µ��Ӳ��ɶԣ�����ƽ�У�������̬������triplet Carbene��

ʵ���ϣ��������ǵ���̬����������̬�����Ľṹ�ǹ��ڼ��ˣ�������ۼ����ʵ��ⶨ�Ľ������������̬�����ļ���Լ103��������̬�����ļ���Ϊ136��
������һ����Ҫ��Ӧ�ǿ�����˫���ӳɵû����黯���������һ�㷴Ӧ����ʽ��

�� Simmons-Smith��Ӧ
���������п-ͭż����п�ۺ�3������Ļ�����У�����2������ͭ��Һ����п�۱������������ͭ���Ӷ�ʹп�ۻ������Ч����ϩ���е�˫���ӳɣ����ɻ������������磺

Simmons-Smith��Ӧ�������������Ե�˳ʽ�ӳɷ�Ӧ��ϩ���Ĺ��ͱ��ֲ��䣺

��6�� Ⱦ�ϼ�飨ż��Ⱦ�ϣ�
Ⱦ����һ�ֿ��Խ��ι̵ĸ�����ά�������ϴ�Ե���ɫ���ʡ����ݻ�ѧ�ṹһ���Ϊż��Ⱦ�ϡ���ϵȾ�ϡ�����Ⱦ�ϡ�������Ⱦ�ϡ����ǰ�Ⱦ�ϡ����Ⱦ�ϡ�̪ݼȾ�Ϻͻ�ͪȾ�ϵȡ�
�� �л�������ķ��ӽṹ����ɫ�Ĺ�ϵ��
1���л�����������й�����ϵ������������ɫ�ļ��������Ϊ������ϵԽ�������ӹ��ԾǨ��������ԽС��Խ����������ˣ������Ⲩ����������

�����Ӿ��е�����ɫ�����յ����ಹ�ġ��������հ���ijһ�ֹ⣬ʣ�¸о�������ɫ������һ��������Ҫ�����ջƹ⣬�ų����Ĺ������ɫ�ġ��磺��ɫ����ɫΪ����ɫ��
����������ɫ�����չ���ɫ�Ĺ�ϵ��
|
������ɫ |
���չ���ɫ�Ͳ���( nm ) |
|
���� |
�� 400-450 |
|
�� |
�� 450-480 |
|
�� |
���� 480-490 |
|
�� |
���� 490-500 |
|
�Ϻ� |
�� 500-560 |
|
�� |
���� 560-580 |
|
�� |
�� 580-600 |
|
���� |
�� 600-650 |
|
���� |
�� 650-750 |
2.���л������ﹲ����ϵ��������ɫ������ɫ��һ���������ɫ�ļ��
�������
�� ��ɫ���ţ���ɫ�ţ���Chromophore�������ۼ�������ԭ�ӻ�����������ӹ����������յģ�һ��Ϊ���������ӵĻ��š�
�磺  �ȡ�
�ȡ�
�� ��ɫ���ţ�Auxochrome��:����ԭ�ӻ��ű�����200nmǰû�����գ�����������ɫ������ʱ����������������շ�IJ�����������ǿ�ȡ�һ��Ϊ����p���ӵ�ԭ�ӻ�ԭ���š��磺  �ȡ�
�ȡ�
��ɫ�������빲����ϵʱ����Щ������δ���õ��ӶԲ��빲����ϵ����������������������ӵ������ԣ�ʹHOMO�ܼ��������������ӣ��Ӷ������˷��ӵļ����ܣ�ʹ�����������������ƶ���������ɫ���

��ɫ�����빲����ϵʱ��ͬ���ܲ��빲�����ã�ʹ������ϵ�����������������ӣ�ʹ���Ӽ����ܽ��ͣ����ղ��������ƶ�����ɫҲ���

��������ϵ���˵��������ŵĵ���ЧӦЭ��ʱ�����ӹ�����ϵ���ȶ��ԣ��Ӷ������յĹⲨ��������

��������ϵ���˵��������ŵĵ���ЧӦ��Э��ʱ���������ƶ�û�ж�������

3.�л�����������ӻ�����ɫ����Ӱ��
���л�������ķ������ӻ�ʱ�����������ȡ�����ĸ��������ƣ�������ȡ������������������ǿʱ������������������ƶ�����ɫ������������ӻ��Ľ�������ӻ��Ĺ������������ͣ������෴��



��������ɫ������Ⱦ���е�����
1. ʹȾ�ϵ���ɫ���
2. ʹȾ�Ͻ��ι̵ع̶�����ά�ϡ�����һЩ�о�����ΪȾɫ��һ�������������������������Ľ�ϡ���
3. ʹȾ�Ͼ���ˮ���ԣ�����Ⱦɫ����  ��
��
������Ⱦ�ϵ�Ⱦɫ���̼���Ӧԭ��
Ⱦ������࣬ż��Ⱦ��������֮һ����������֪Ⱦ���У�ż��������Լռ�������ϡ��ڷ�֯��ҵӦ���ϣ����ǰ����˼��ԡ����ԡ�ֱ��ýȾ�ͱ�ȾȾ�ϵȼ����ࡣ�����Ա�ȾȾ���з�������Ϊ��˵����ȾȾ�ϵ�Ⱦɫ���̼���Ӧԭ����
������������������ˮ�����Կɰ���ȾȾ�Ϸ���Ⱦɫ��Ⱦɫʱ���Ƚ�֯���÷�������ν��ϳɷ����������õķ����Ʒ���AS����Ȼ�����ý��зӵ�֯��ͨ��ɫ���������������ص���������Һ�ϳɷ����������䷴Ӧ���£�

��3 ��������
����������ָ���Լ������Ȼ��������ص�Ӱ���£�������ijЩԭ�ӻ���ŷ���ת�ƻ����̼��ı������µ����ʵķ�Ӧ��
������������˺������ź���������Ž��м�Ҫ�����ۡ�
һ��������
������������1��2��λ�ƣ����ڷ�Ӧ�У�һ��ԭ�ӻ���Ŵ���һ�Ե���ת�Ƶ����ڵ�ȱ���ӵ�ԭ���ϡ�

1�� ���ŵ�ȱ���ӵ�̼ԭ����

��1��1�� ���������ӵ����ţ�


��2��Ƭ�Ŵ�����



������ȡ���Ҷ�����ͳ��Ƭ�Ŵ��ࣩ��ǿ�������¶��ᷢ��Ƭ�Ŵ����ţ��ṹ��������ͨʽ��ʾ��

����R������ͬҲ���Բ���ͬ�����������Ҳ�����Ƿ�������R����ͬʱ����������Ƭ�Ŵ����ŷ����Ԥ�����ŵIJ����Ϳɿ��ǣ�
�� �������ǻ��У���һ���ǻ����ȱ����ӻ��γ����Σ�����̼�����ӣ��������ȶ�̼�����ӵ�̼�ϵ��ǻ����ȱ����ӻ���

�� ����̼�����Ӻ����ڵ�������������һ�����ȷ���ת�ơ�
����ת��˳��ͨ���ǣ�����>�����
��ǿ�ģ������ܶȸߵ���ת�ơ�ת�ƻ��ŵĵ����ܶ�Խ�ߣ�����Խ��Խ��ת�ơ�

������̼������֮�����ڵ�������������һ��ת�ƣ�һ���˵�����ռ�λ�費��ʱ����Ҫ�����������������˵����ƣ������ƴ�ķ�������ת�ơ����㻷�ĵ����ܶ�Խ�ߣ���ת�Ƶ�����Խ��


����̼�������γɻ������ȶ��м���Ļ�������ת�ơ��ڼ�������ת������ϵͣ�������ǿռ�λ���ԭ��
�� Wagner��Meerwein���ţ�
��ß��ȡ�����磺

����ˮ�����Լ����磺HX��PCl5��SOCl2��������±����ӳɵ��ϩ�Լ��±��������±����ʱ����ʱ������������̼�������š����෴Ӧ��1899�걻Wagner���֡�����Meerwein���Է�չ�������ΪWagner��Meerwein���š�
Wagner��Meerwein����Ҳ��ͨ��̼�������м�������ɶ����еġ�
��1��


�ϩ��������Ĵ�����Ҳ����Wagner��Meerwein���ţ�����a����ϩ
��2��


��Ȼʱ����̼����������Ϊ�ڶ�̼�����ӣ������ɺ���Ԫ����̼���������ŵ���Ԫ����̼������֮��������С�ˡ�
��3��

��4��

���Ƿ�����һ��̼�����м��壿�����ȿ�һ��ʵ����ʵ��

�������ص��ηֽ�����̼�����ӣ���ô����ӦΪC6H5CH2C*H2OH����ˣ���Ϊ��Ӧ�Ǿ���������ʾ���м��塣
���ԣ���Ӧ���Ǿ���̼�������м��������м���

RΪ��ϩ��ͬ�������������м��壺

���DZ�������ϩ��������λ������ţ����ǵ������������ȶ����ڵ�̼�����ӵ�Ե�ʡ�
��5��

��Ӧ���̣�


��4�����ŷ�Ӧ�е����廯ѧ
�����ŷ�Ӧ�У����Ǩ�ƻ��ű�����һ�����Ի�����ôǨ�ƺ��乹���Ƿ����仯��Ǩ������̼ԭ�Ӻ�Ǩ�Ƶ�����յ�̼ԭ�ӣ����ǵĹ����Ƿ����仯��Ǩ�ƻ��ź���ȥ�����ڿռ�λ������ʲô��ϵ����Щ�����������ŷ�Ӧ�����廯ѧ��Ū����Щ���⣬����Ԥ����
�Ų��K�乹���Ǻ���Ҫ�ġ�
�� ʵ��֤���������ŷ�Ӧ1��2��Ǩ���У�Ǩ�ƻ��ŵĹ���û�иı䡣����Դӹ���̬�ķ��ӹ��ģ�������⣺

��ʼʱ��Ǩ�ƻ�����CR1R2R3��C2��sp3��sp3�ӻ������ص��γ����������ڹ���̬����CR1R2R3��C2�ɼ�����������Ϊ����ԭ�ӹ��ã���CR1R2R3���ŵ�sp3�����C1��C2��p���������γ���һ�������Ķ����ӵĹ���̬��Ȼ����CR1R2R3��C2���ѣ��γɣ� II ����
�� ʵ��֤��Ǩ�Ƶ���ʼ����յ��̼ԭ�Ӷ��й����ϵķ�ת



�� ���ŵ����廯ѧ�о�˵���������Ź�����ת�ƵĻ��ź��뿪�Ļ��ű˴˴��ڷ�ʽ��


���ŷ�Ӧ���л��ϳ��е�Ӧ�ã�
��1

���̣�


��2�������۵�ԭ�Ϻϳ�3��3��������2������


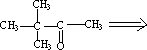

��3���ӻ���ͪ�ϳɻ���ͪ��

�ϳɣ�

2.���ŵ�ȱ���ӵĵ�ԭ��
(1)Beckmann����

ȩ��ͪ����������������Ϊ�������ⷴӦ��ΪBeckmann���ţ��䷴Ӧ���̱�ʾ���£�

��

Beckmann�����Ǿ���������һ�����������м��壬�뵪���������ڵ����������ǻ����ڷ�ʽ��ת�Ƶ���ԭ���ϣ������γ�̼�����ӣ�ˮ���Ӽӵ�̼�����������������Ӻ�ǿ����OH2����Ȼ����ȥ�������ų�������
�磺����[4��3��0]��ͪ��7��뿵�E��Z�칹�����ŵò�ͬ�IJ��


���ǻ����ڷ�ʽ������ת�Ƶ���ԭ���ϡ�
����ͪ�Beckmann���ź��ɼ���������
���Ǻϳɾۼ��������ĵ��塣
Beckmann���Ÿ������ṩ����ȩ��ͪ������ȡ�����ķ�����
��ҵ��

��2�� Hofmann����
��ͼ��ˮ��Һ����������ʱ���ų�������̼��������һ��̼ԭ�ӵİ���

�����Ʊ�������һ����Ҫ����������Hofmann���⡣��Ӧ���̱�ʾ���£�

�����һ���γɵ�N��±�����������������õ����ڵڶ�����±������������ȡ��һ�����ӣ�±�������ĵ�ԭ�������������������ӻ��ţ���������Եģ��������������������̣��ѵ����γ��˴�����ϩ����R����ת�ƣ������϶�����������������Эͬ�ġ�
����һ�ֹ۵���Ϊ��


��������̼ԭ�ӵ�������������������Hofmann���ŷ�Ӧ�����������ŷ�Ӧ��������
��95.5 %��ѧ���ȣ��������������ͱ��ֲ��䣩��

��˵��Ǩ�ƻ���û��������ӣ����Ǩ�ƻ��ŵĹ��ͱ��ֲ��䡣
��ҵ��

3.Wolff���ţ�̼ϩ�����ţ�
�����ص�ͪ��Ag2O�����¹��ջ����ʧȥN2�����ŵõ���Ӧ�Լ��ߵ�ϩͪ���ⷴӦ��ΪWolff���ŷ�Ӧ��

4.���ŵ�ȱ���ӵ���ԭ�ӣ�
(1)����������������



(2) Baeyer��Villiger����

�ڹ����������������£�ͪת��Ϊ��Ӧ������


����ת�Ƶ�һ��˳��Ϊ��H>C6H5- >�����>�����>����� >����
�������ͪ�������������������ǽϴ����Ǩ�ƵĽ��������ת���������ŵ���ȱ��������һ�ձ�ԭ��Ǩ�ƻ��ŵĹ����ڰݶ���ά����������Ӧ�б��ֲ��䡣���������롶�ߵ��л���ѧ��P298~299��


����������
ǰ�������۵���������ָ��������ȥһ�������ӣ�����һ��̼�����ӻ�����ȱ���ӵ���
��,������֮���ڵĻ��Ŵ���һ�Ե���ת�ƹ������෴,�����ŷ�Ӧ���ڷ�������ȥһ��
������,����һ��̼�����ӻ���л��õ�δ���õ��ӶԵ�����,����֮���ڵĻ�����������
����ʽת�ƹ���,��ת�ƻ�����������һ�Ե���,������ȡһ������.

�����ŷ�Ӧ����,�������Stevens����Ϊ��˵������,���л��õ���-��ԭ�ӵļ����
�����,��ǿ����������-��ԭ����������ʽ�����γ�̼������,�����ӵ�ԭ�ӻ���ԭ��
ת�Ƶ����ڵĸ�̼������,��һ��Ӧ��Stevens���˷���,���Գ�ΪStevens����.

�������е�ԭ�ӻ���ԭ�ӵ�����ɺ��ʻ�ʹ�Ǽ��,�������ɸ�̼���ӡ�Ǩ�ƻ��ų�Ϊ
�л���ϩ����.
ʵ��֤���ڷ�Ӧ������Ǩ�ƻ��ŵĹ��Ͳ���.

˵��C-N�������Ѻ�C-C����������Эͬ���е�.
��.���������
�����������ָ�����������, ������������ŵķ�Ӧ. ����������о���������
1,2-����ת��,��һ��������һ��λ��ת�Ƶ����ڵ�λ����ȥ.
����,��-��������ȩ,���ȱ����ж��嶡����������(di-t-butyl peroxide)������
�γɴ�Լ������������Ʒph-CMe3�ͷ����ɱ���ת���γɵ����Ų�Ʒ.






ֵ��ע�������������ŵ��ȱ�̼������С�Ķࡣ������ʵ����ֻ��50%ת�ơ��پ�
������û�м�ת��,һ���ڳ�������²��������������ת��.����������,��ϩ��
������������±��Ҳ��ת�ơ�
��.����������

�����������,��Xȡ����������ԭ�ӻ�ԭ����,ת�Ƶ���������λ���λ��
X�ǵ�����,��Y��ǿ�������ӻ���,�����ŷ�Ӧ�����������������ŷ�Ӧ�漰������,���Գ�Ϊ�������š�
1.N-�ǻ�����������:


2.����������
�⻯ż������ǿ������,�������ŷ�Ӧ����������

���������ſ�����ͨ�����������ɻ����е�:




3.Claisen����
Claisen��1912�귢��,����ϩ�����Ѽ�����200��ʱ,ϩ��������ԭ��ת�Ƶ���������λ
.��������λ������ȡ����,��ת�Ƶ���λ,����ϩ������:



4. ����˹(Fries)����

һ���ڵ��������ɶ�λ�칹��,�ڽϸ��¶���������λ�칹�塣
Fries ���ŵ����̻�ʮ�����,��λ�칹��������ɷ����ڵ�������Ӧ���ɵġ���λ�칹������Ƿ������Ų��

�ο���:���л���ѧ���²� �Ͼ���ѧ�� P166��188
�������л����²� ������ P315--334
5������������������(Benzilic acid rearrangement)

�� ������������


6��Favorskii rearrangement