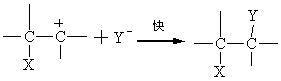
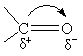
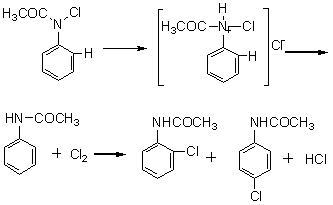
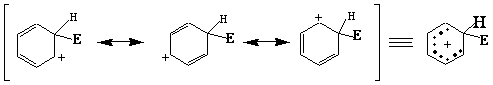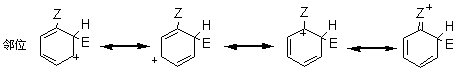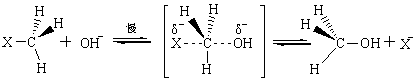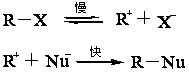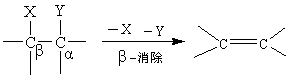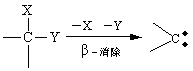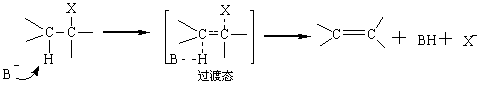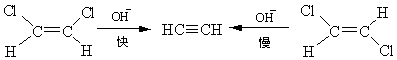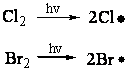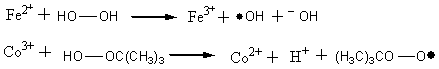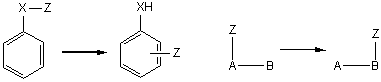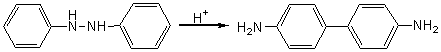|
��ϸ�л���ѧƷ�ĺϳ��빤��ѧ �ڶ��� ��ϸ�л��ϳɵ����ۻ���
���ߣ����� ת���ԣ���վԭ�� �������143
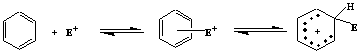
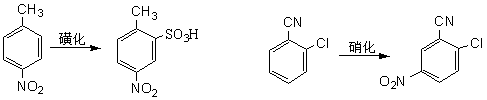
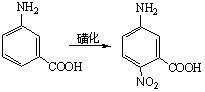
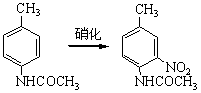
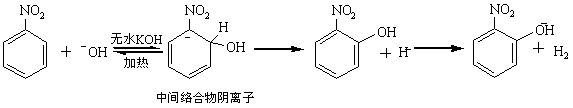
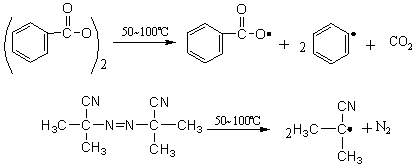
�ڶ��� ��ϸ�л��ϳɵ����ۻ��� ��ϸ�л��ϳɷ�Ӧ���ս��з�ʽ��ͬ������ʽ�Ͽ��Է�Ϊȡ����Ӧ���ӳɷ�Ӧ��������Ӧ���Լ��������ŷ�Ӧ�ȡ�ÿһ�ַ�Ӧ�ֿ��Է�Ϊ�������ࡣ ȡ����Ӧ���ݷ�Ӧ�Լ����ʺͷ�Ӧ�������̼-������ѷ�ʽ��ͬ����Ϊ��ȡ������ȡ���������ȡ����Ӧ���ӳɷ�Ӧ���ݼӳɵĻ���;����ͬ�����Է�Ϊ��ӳɡ��˼ӳɡ�������ӳɺͻ��ӳɡ�������Ӧ���Ը��ݱ�����ԭ�ӻ�ԭ����λ�ò�ͬ����Ϊ��-��������-�����ȡ����ŷ�ӦҲ���Է�Ϊ�����ࡣ 2.1 ��Ӧ�Լ��ķ��� 2.1.1 �����Լ� �����Լ���ָ��Щ�ܹ����������һ�Ե������γɹ��ۼ����Լ��������Լ��ַ�Ϊ���Լ������Լ��� 2.1.1.1 ���Լ� ���Լ��Ǵӻ�����ȡ��һ�Ե����γɹ��ۼ����Լ��������Լ��������ܶȽϵͣ�Ӧ�н����������ӵĸߵ������ܶ����ģ����������ܣ��������¼��ࣺ �����ӣ�NO2+��NO+��R+��R-C+=O��A rN2+��R4N+�ȣ� ���пɼ������Ѿ��������ۼ��ķ��ӣ�Cl2��Br2��HF��HCl��SO3��RCOCl��CO2�ȣ� ���пɽ��ܹ��õ��ӶԵķ��ӣ�δ���ͼ۵��Ӳ�ԭ�ӵķ��ӣ��� AlCl3��FeCl3��BF3�ȣ� �ʻ���˫���� ��������Fe3+��O3��H2O2�ȡ� ���ࡣ ±�����е������R-X�� �ɸ����Լ�������������ӷ�Ӧ���練Ӧ�����磺��ȡ������ӳɡ� 2.1.1.2 ���Լ� ��һ�Ե����ṩ���������γɹ��ۼ����Լ������Լ��������Լ����нϸߵĵ������ܶȣ���������������ʱ���������÷��ӵĵ͵������ܶ����ģ����������ܣ��������¼��ࣺ �����ӣ�0H����RO����ArO����NaSO����NaS����CN���ȡ� ���Է�����ż���ĸ��ˣ�NH3��RNH2��RR`NH��ArNH��NH2OH�ȣ� ϩ��˫���ͷ�����CH2��CH2��C6H6�� ��ԭ����Fe2+�������ȡ� ���ࡣ �л������������е������RMgX��RC��CM�� �ɸ����Լ�������������ӷ�Ӧ���˷�Ӧ�����磬��ȡ�������û����˼ӳɵȡ� 2.1.2 ������Լ� ����δ�ɶԵ����ӵ������������һ�������¿ɲ���������Ļ������������Լ������磬�ȷ��ӣ�C12���ɲ������������C1������ 2.2 ��ȡ����Ӧ ��ϸ�л��ϳ��е���ȡ����ӦҲ�ɳ�Ϊ��������ȡ����Ӧ�����㻷��һ����״������ϵ�����ڻ�����-�������ܶȽϸߣ���������ȡ����Ӧ�� 2.2.1 ��������-���������-����� �������к�һϵ�����Լ��γ����������ԡ�ͬ�������������Լ��γ��������뷼��ƽ������Ļ�״��-�����Ʒ�����ɢ��ϣ����ʵ�ͬ������̼ԭ��֮��û���γ������Ļ�ѧ���� ����������ǿ���Լ�ͬ�������ڷ�Ӧ˲���ܴӷ����϶�ȡһ�Ե��ӣ��뻷��ijһ�ض���̼ԭ���γ���-�����γ���-������Ʒ������ӣ�����-��������-�����֮�������ƽ�⣬��-������Ϊ�ȶ�����ij������£��ܽ������õ������磬���������顢����������������������100��ʱ����һ����ɫ�Ľᾧ̬��������-������������50���������ȶ��ģ���������50����ֽ�ɼ����������ױ������������������
�Ѿ��ж����о��������֤�����������ȡ����Ӧ�ǰ��վ�����-������м�����������̽��еġ������ʵ�E+�Ľ��������ӵ�����ͬʱ������һ�����̣���һֱû�з��ֹ������������ʵ����������ǰ�����Ӿ��Ѿ����������ĵ��������̣�ֻ�ڼ����������²����С�
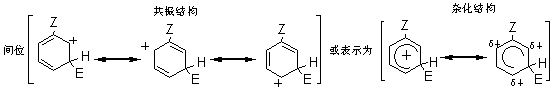
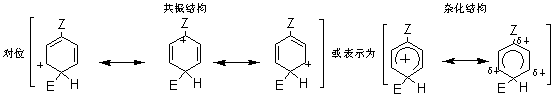
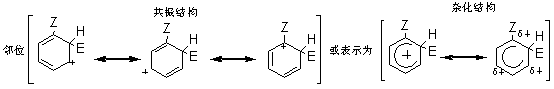
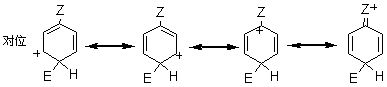
2.2.2.1 ����ѧͬλ��ЧӦ �����κη�Ӧ����ν������ѧͬλ��ЧӦ����ָ�������Ӧ������е�ijһԭ��������ͬλ�ش���ʱ���÷�Ӧ�ٶ��������ı仯�����磬�������ͬλ����H���D���T����������ͬ��������ɵ�̼��������ٶ����в��ģ���������Ķ��ѽ������ݾ�ʵ�����ݣ�C-H���Ķ����ٶ�Լ��C-D����7����Լ��C-T����20��������1�������������̶����ٶȿ��Ʋ�����H+�����䣨��k2<k1������2������һ�����̻��ߣ�3�����յ��������̣���ô����ͬλ��ЧӦkH/kD�����ӽ���7������kH/kT�����ӽ���20���������������̶����ٶȿ��Ʋ�������-���������ɣ���û��ͬλ��ЧӦ�� 2.2.2.2 ��-�����ķ����������ȶ��� ��Ӧ��-����������ǿ��Ʋ���ʱ����һ�����ɾͿ��ٵ������Ӷ�ת��Ϊ���һ�㲻�ܰ����Ƿ��������Ҳ���۲쵽���ǵĴ��ڡ�����ijЩ��������£����ܷ�����м������-�������磬���������顢����������������������100��ʱ���ɵĻ�ɫ�ᾧ̬�������Ǹ��������м������-����Ȼ���ٷֽ��γɲ�������������ױ��� ������ȡ����Ӧ������������Լ��������㻷�������γ���-����Ȼ��ת��Ϊ��-��������������ˮҺ̬������ʱ��������-��������-������ƽ�⡣ 2.2.3 ��������ȡ����λ���� 2.2.3.1 Ӱ�춨λ����Ҫ���� ����������һ����ȡ����������������ȡ����ʱ��������λ�úͷ�Ӧ���е��ٶȣ���Ҫȡ�����������ء� ��1������ȡ���������ʣ���������ЧӦ�Ϳռ�ЧӦ��������м���ȡ��������������ǵ����ʺ����λ�á� ��2�����Լ������ʣ�Ҳ��������ЧӦ�Ϳռ�ЧӦ�� ��3����Ӧ������Ҫ���¶ȡ��������ܼ���Ӱ�졣 �����������У�����Ҫ��������ȡ�����ļ���ЧӦ���ڷ���ϵȡ����Ӧ��,��ϵ����ȡ���о�����࣬Ҳ����Ҫ�� 2.2.3.2 ���ඨλ�� ����ȡ���У�����������ȡ��������ȡ�����Ķ�λ�������������ͣ����ڡ���λ��λ�ͼ�λ��λ��ͨ�����ڡ���λ��λ��������һ�ඨλ�����Ѽ�λ��λ�������ڶ��ඨλ���� ���ڵ�һ�ඨλ������Ҫ�У� ��O-����N(CH3)2����NH2����OH����OCH3����NHCOCH3����OCOCH3����F����C1����Br����I����CH3����CH2Cl2����CH2COOH����CH2F�ȡ� ���ڵڶ��ඨλ������Ҫ�У� ��N(CH3)2����CF3����NO2����CN����SO3H����COOH����CHO����COOCH3����COCH3����CONH2����N+H3����CCl3�ȣ� ������ν�ڡ���λ��λ���λ��λ�����ǶԷ�Ӧ����Ҫ������ԡ� 2.2.5.3 �����Ķ�λ���� 1.����ȡ�����ļ���ЧӦ�ڲ�������ȡ���У����Ը��ݱ���������ȡ�����ļ���ЧӦ�������ɸ��칹��Ħ�-�������ȶ��������Ͷ�λ���á���һȡ�������ȡ�����������ڡ��Ժͼ�λ�����֦�-����ÿһ������ﶼ���Կ��������ֹ���ṹ�ӻ��Ľ��������λ�Ͷ�λ��������У�����һ������ṹ��������ɼ�����ͬ����ȡ����Z������̼ԭ���ϡ�������ӻ��ṹ�У�ͬZ������̼ԭ���Ͼ��в�������ɡ��ڼ�λ��������У���������ṹ��ͬZ������̼ԭ���϶�û������ɼ��С�����������ӻ��ṹ�У�ͬZ������̼ԭ����û�в�������ɡ����ֲ���ǽ�������ȡ������λ���õĻ�����
��Z�ͱ���֮���й���ЧӦʱ��ijЩ�����ͬ�յ�ЧӦ�ķ���һ�£�����һЩ�������ͬ�յ�ЧӦ�ķ����෴��һ����˵������ЧӦ���������á���Z��ͬ����������ԭ�Ӿ���δ���е��Ӷ�ʱ������δ���е��ӶԷ�ɢ�������ϣ�ʹ������ȶ��������Ƕ�����λ�Ͷ�λ���������Զ��һ������ɼ�����Z�ϵĹ���ṹ�� �������Ĺ���ṹ����������ṹ���ȶ��������ӻ��ṹ����λ�Ͷ�λ�������ȶ����������ȡ����ʹ��������������ڡ���λ��λ��
�����������ۣ�����ȡ�����ɹ���Ϊ�������ࣺ ��1��ȡ����ֻ�������յ�ЧӦ��������������Ƕ�ʹ��������������ڡ���λ��λ�����м������г�����ЧӦ�������ô������������ ��2��ȡ������ͬ����������ԭ�Ӿ���δ���е��Ӷԣ����磺��O�C����NR2����NHR����NH2����OH����OR����NHCOR����OCOR����F����C1����Br����I�ȣ���δ���е��ӶԺͱ����γ����Ĺ���ЧӦ��+I�������Ƕ����ڡ���λ��λ��������������ЧӦ�⣬��Щȡ����Ҳ�������յ�ЧӦ�����С�O�CΪ���յ���+I���������Ķ����и����յ�ЧӦ�����ڰ������ǻ��������Ĺ���ЧӦ��+I�����ڸ����յ�ЧӦ��-I�����������Ƕ�ʹ�����������±�أ������Ĺ���ЧӦС�ڸ����յ�ЧӦ������ʹ�������Զۻ��� ��3��ȡ�������и����յ�ЧӦ������ͬ����������ԭ��û��δ���е��Ӷԣ����磺��N+R3����NO2����CF3����CN����SO3H����CHO����COR����COOR����CONH2����CCl3���͡�NH3�ȣ�����ijЩ���յ�ЧӦ�⣬���и��Ĺ���ЧӦ�����Ƕ�ʹ�����ۻ��������Ǽ�λ��λ�� 2.����ȡ�����Ŀռ�ЧӦ ����������ȡ�����Ŀռ�ЧӦ����ָ���ǿռ��ϰ����á��������һ����ʱ����������������λ�칹����ı�����С�� 3. ���Լ��ļ���ЧӦ ���ʵ�E+�Ļ����ԶԶ�λ����Ҳ����ҪӰ�졣��E+������ʱ��kT/kBֵС����E+�����ױ����������ѡ���Ժܲͬ�������ױ����ϲ�ͬλ�õ�ѡ����Ҳ�ܲ��������൱�����ļ�λ�칹�������ױ���C-�黯�ͽӽ����������˵����CH3����CH2CH3����������Ӳ��Ƿdz����õ����ʵ㡣 ��֮����E+��������ʱ���������ױ��ͱ���ѡ���Ժܺã�kT/kB��Ҫ�����ڼױ��ͱ�����Ի��ԣ�����Ҫ�����ڼ��Ļ���ã����kT/kB�ܴ�ͬ����E+�����ױ�����ͬλ�õ�ѡ����Ҳ�ܺã����������ɼ�λ�칹�������ױ���±����C-�����ͽӽ����������˵������̬�ȣ�Cl2���͡�COCH3�Ǻ��������ʵ㡣 4.��ȡ�����Ŀռ�ЧӦ ��ȡ�����Ŀռ��ϰ�Ҳ��Ӱ����λ�칹��������ɱ����� 5.��Ӧ������Ӱ�� ��1���¶ȵ�Ӱ�� �¶����߿���ʹ������Ļǻ���C-�黯ת��Ϊ���淴Ӧ���¶ȵı仯�Բ�������ȡ�����칹�������Ҳ��Ӱ�졣��������������������Ӧ�����������¶ȣ��������������������ɱ������½��� ��2��������Ӱ�� �������Ըı����Լ��ļ���ЧӦ��ռ�ЧӦ������ױ��û�������ʱ������һ�������ᣬ������߶�λ�칹������ʣ�����λ���������36%��ߵ�40%���������������-NO2���ɵ��������Ϊ�����ʵ㣬�������ʹ��λ�칹������٣���λ�����ӡ�����Ҳ���Ըı䷴Ӧ���̣��������Ļǻ���Hg��ʱ���ǻ�������λ����Hg�ν�����λ�� ��3�����ʵ�Ӱ�� ��Ҫ�ǽ�����ȵ�Ӱ�죬���磬���������������������������������������ܵõ��������λ�칹��� 6. ��������ȡ�����Ķ�λ���� ����������������ȡ���������������ȡ����ʱ����ȡ�������뻷��λ����Ҫ����������ȡ���������͡���λ������ǿ�������ǵ����λ�á�һ��������ȡ������λ����һ�ºͲ�һ�����������
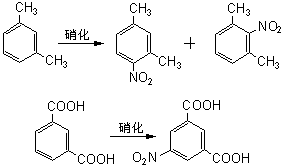
������ȡ��������ͬһ���ͣ������ڵ�һ���ڶ��ඨλ�����������ڼ�λ���䶨λ������һ�µģ����磺 �ɼ���ȡ�������ٽ����������ڼ�λ��ȡ����֮�䡣��Ȼ���ǿռ�ЧӦ�Ľ������������ȡ����������ʵ������������������ԡ� ������ȡ�������ڲ�ͬ���ͣ���������λ���λʱ���䶨λ����Ҳ��һ�µġ����磺
��������Ѵ�ȡ��������ȡ�����Ķ�λ���ò�һ�£���ȡ���������λ�ý�����������ȡ��������Զ�λ������ͨ����һ��ȡ�����Ķ�λ�����ȵڶ���ǿ�öࡣ
������ȡ�������ڲ�ͬ���Ͳ����ڼ�λ���䶨λ���þ��Dz�һ�µģ���ʱ��ȡ������Ҫ�����һ��ȡ��������λ���λ�����磺 ������ȡ��������ͬһ���Ͳ�������λ���λ������ȡ���������λ�þ����ڶ�λ������ǿ��ȡ���������磺
2.3��ȡ����Ӧ ��ϸ�л��ϳ��е���ȡ��Ҳ�ɳ�Ϊ��������ȡ����Ӧ�������Լ������ʺͷ�Ӧ�������C-H���Ķ��ѷ�ʽ���ɰ����·�Ӧͨʽ��ʾ��
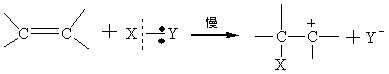
��Ӧ�Ȱ�����������ȡ��Ҳ����֬�������ȡ������Ӧ�ý϶��Ϊ֬�������ȡ����Ӧ�������������ҪӦ�������û���Ӧ�� 2.3.1 ֬������ȡ����Ӧ���� �ڱ���̼ԭ���ϵ���ȡ����Ӧ������͵ķ�Ӧ��±������������Լ���������ȡ������SN��ʾ���䷴Ӧ������SNl��SN2������ʽ���ֱַ��������£� 2.3.1.1 ˫��������(SN2) SN2��ʾ˫������ȡ������������оɵĻ�ѧ�����Ѻ��µĻ�ѧ���γ���ͬʱ�ģ����м�������ɣ���Ӧͬ�����С��Բ�±���鷴ӦΪ����
���Լ��Ľ����ӱ�������ȥ����180���λ�ýӽ����������̼ԭ���γɽ����ļ���ͬʱ��ȥ����̼ԭ�ӵ����Ӽ�����������̼ԭ�ӳ�һ��ֱ�ߣ���̼ԭ����������������ͬһƽ���ڡ���������̬�γɡ���һ���̽��н������Ƿ�Ӧ�Ŀ��Ʋ��衣����Ӧ�ɹ���̬ת���ɲ���ʱ��̼ԭ����������������ƽ������һ�߷�ת�����ò���Ĺ�����ԭ��������෴����Ϊ�ڿ��Ʒ�Ӧ�ٶ�һ����������������Ӳμӣ����Խ�˫������ȡ�����ڷ�Ӧ�з����˷��ӵĹ�����ת����ȥ������Ϊ���߶���ת�������ڷ�Ӧ�У�������������ת����ΪSN2�ͷ�Ӧ����Ҫ��־�� 2.3.1.2 ���������̣�SN1�� SN1��ʾ��������ȡ�����������Ӧ�����з���������ȡ����Ӧ����һ������ȥ��������̼ԭ��֮��ļ��������ѣ�����һ�����ȶ���̼�����ӣ���һ�������裬�ڶ����������������̼�������м��壬Ѹ�������Լ���Ϲ����¼���
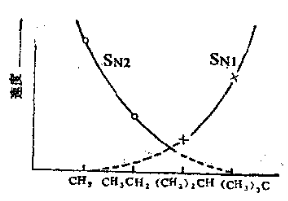
������Ӧ�ٶȾ����ڵ�һ���������̡����ڸò�����ֻ��һ����������Ӳμӣ����Խ�����������ȡ�����������������ȶ���Խ�������R-X�������������̽��з�Ӧ������Խ�÷�Ӧ���������ǵ����ӵģ�һ�������������-̼ԭ���ϻ��ۣ����۴ӵ���ЧӦ���ǿռ�ЧӦ���������Լ��Ľ������һ�����ѣ�ʹ��SN2���̵ķ�Ӧ�ٶȽ��ͣ�����������-̼ԭ���ϵ������ܶ�����±ԭ�Ӿͽ����׳�Ϊ�����Ӷ���ȥ�����ɵ�������������ڳ�����ЧӦ���ڣ��нϴ���ȶ��ԣ���Щ���ض���ʹ��±���ˮ�ⷴӦ��SN1���̽��С� 2.3.2 ��Ӧ��Ӱ������ Ӱ����ȡ����Ӧ���̺��ٶȵ����أ���Ҫ��������Ľṹ�����Լ�����ȥ���ź��ܼ������ʵȣ���������֮�������ϵ�ġ� 2.3.2.1 ������ṹ��Ӱ�� ������ṹ��SN1��SN2��Ӧ���ȵ�Ӱ���е���ЧӦ�Ϳռ�ЧӦ�������ء�����±�����ˮ�ⷴӦ�������SN2���̽��У��� ��±�飾��±�飾��±�� ����Է�Ӧ���ʴӲ���������ﵽ������������Լ��С103�����������������̽������� ��±�飾��±�飾��±�� ����Է�Ӧ�ٶȴ�Լ���106�������Ͳ�����������ǰ�SN1���̽��з�Ӧ������������ﰴSN1���̽��з�Ӧ������������������SN1��SN2�ı߽�״̬����������ͼ��ʾ��
±���������ˮ���ٶȵ�Ӱ�� ������������У���������̼ԭ���������������Ի��š�����-���ѡ�3-�ȱ�ϩ�ȡ�����ȡ������p-p�����p-������ЧӦ��ʹ���ɵ�̼�������ȶ������ǵķ�Ӧ�ٶȱ�û��ȡ�����Ĵ���ǧ�������������䷴Ӧ��SN1���̽��С� �ڱ�������̼ԭ������������ȡ����������-±���ʻ��������-±�����������ȡ�����ȡ���������������ã�ʹ��������̼ԭ�ӵIJ�����������ӣ�������SN2��Ӧ��ͬʱ�����γɹ���̬ʱ��ȡ��������-������Ҳ���������γɼ������ڶ��Ѽ��ĵ����ƽ��ǣ�ʹ����״̬�������ͣ�ʹSN2�ķ�Ӧ�ٶ����ӡ� ��֮������������У���������̼ԭ�����и�����ȡ������������SNl��Ӧ����������������ȡ��������������SN2��Ӧ�� 2.3.2.2 ��ȡ����ȥ���ŵ�Ӱ�� ������SN1��SN2�ķ�Ӧ�У���ȡ���Ļ���X���Ǵ���ԭ�����е�һ�Ե�����ȥ������X���ܵ�������ԽǿԽ����ȥ��ҲԽ��������ȡ����Ӧ�Ľ��С�һ��˵�����䱻ȡ�������״���Ϊ�� RSO3���� I���� Br���� Cl���� RCOO���� HO���� H2N�� 2.3.2.3 ���Լ���Ӱ�� ��SN1��Ӧ�У����Լ������ʶԷ�Ӧ�ٶ�û��Ӱ�죬��Ϊ���Լ�������������Ӧ���̵��ٶȿ��Ʋ��衣��SN2�ķ�Ӧ�У����Լ������˹���̬���γɣ������������ı仯��ȡ����Ӧ�ٶ�������Ӱ�졣��������Լ���������������Ե�ǿ����һ�µġ����磺�������Լ����ô���Ϊ�� C2H5O���� OH���� PhO���� CH3CH2����H2O ��ͬ��Ԫ�ص��Լ��У������ǰ��縺�Ե��½�����ߡ����磺 I����Br����Cl����F���� PhS����PhO�� ����ԭ������Խ��Խ�����������Ը���������ҲԽ�� 2.3.2.4 �ܼ���Ӱ�� �ܼ��ļ��Զ���ȡ����Ӧ�����ͷ�Ӧ�ٶȶ��кܴ�Ӱ�졣����SNl�ķ�Ӧ����Ӧ�ĵ�һ����һ�����Ի��������Ϊ�������е�ɵ����ӣ���˼����ܼ�������Ӧ���С������ܼ�����Խ��Խʹ��Ӧ�ٶȼӿ졣SN2�ķ�Ӧ�������ܼ������Լ������γ����ʹ���Լ������Լ������ڷ�Ӧ���Լ��뷴Ӧ���γɹ���̬�����ȵ�����һ���������ƻ����ɵ���������Է�Ӧ�ڲ��γ�������ܼ��н�����������Ӧ�ٶȿ졣��֮SNl��Ӧ���������ܼ��н�����������SN2��Ӧ�ڷ��������ܼ��н��������� ֬������ȡ����Ӧ�ھ�ϸ�л��ϳ��н�Ϊ���ã��㷺����̼���¼���̼-̼�¼����γɡ��������ڴ����Ѻ�����ĺϳɣ���±���������ܼ��⣬�γ�̼-���¼���������ˮΪ�ܼ����ɴ����ô�Ϊ�ܼ������ѣ�������Ϊ�ܼ������������������������ѵĺϳ��γ�̼-���¼�������±�����İ����γ�̼-���¼������Ǻϳ��鰷����Ҫ��������ȡ����Ӧ�����γ�̼-̼�¼���Ϊ��Ҫ����Ӧ����̼��������Ϊ���Լ�����̼��������˵��ʹ�����������������ֳ���������Ӧ�����Ժϳ������Ȳ�������̼������Ϊϩ�������ӣ�������������Ӧ�Ǻϳ�ͪ�����ᡢ����������Ȼ��������Ҫ�ϳɷ����� 2.3.3 �����廷�������ȡ����Ӧ �ڸ��෴Ӧ�У����Լ����Ƚ��������ϵ������ܶ���͵�λ�ã������ڷ�Ӧ������λ���ɷ��涼�뷼������ȡ����Ӧ�෴�� �����������ǻ���Ϊ����������ȡ����Ӧ���̼�ʾ���£�
2.4 ������Ӧ ������Ӧ��ָ�л��������ͬʱ��ȥ����ԭ�ӣ�����ţ����γ�һ���·��ӣ�ͨ���Dz����ͳ̶����ӵķ�Ӧ�����ڱ���ȥ������ԭ�ӣ�����ţ���λ�ò�ͬ��������Ӧ��Ҫ�����֣�����-��������-������ ��-����������ϩ����Ȳ����������ķ�Ӧ���������ڵ�����̼ԭ���ϳ�ȥ�������š�
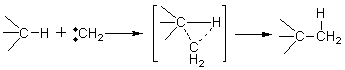
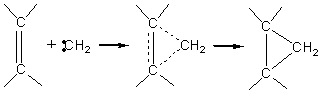
��-���������γ�̼-̼˫����Ҳ���γ�̼-��˫����̼-��˫����̼-��˫���ȡ� ��-���������ɿ�����Carbene���ķ�Ӧ����ͬһ��̼ԭ���ϳ�ȥ�������ţ�Ҳ��1,1-������
2.4.1 ��-������Ӧ ͨ���о�������Ӧ�����̣��ɷ�˫�������̣�E2���͵��������̣�E1���� 2.4.1.1 ˫����������Ӧ���̣�E2���̣� ˫����������Ӧͨ����ǿ�����Լ������·����������Եļ����Լ�B�ӽ�����ʱ����B��H���γ�������ͬʱ��ԭ��C-H����C-X���������γɹ���̬��������C-B����C-X��ͬʱ���ѹ���ϩ����
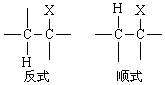
�ɼ�E2���̺�SN2���̺����ơ�����������E2�����м����Լ�������-��ԭ�ӣ�����SN2�����з�Ӧ��������-̼ԭ���ϡ������ڲ�����̼ԭ���ϵ���ȡ��������������Ӧ������ ��E2���̽��з�Ӧ����ȥ���ŵĿռ��Ų��������������֣���˳ʽ�����ͷ�ʽ������
��������������-������������ת������ȷ�������ַ�ʽ���С� ��ϩ���������֬���������У�˫���ͻ��ϵĵ���������ת�ܵ��谭������һ��������ʽΪ������1,2-������ϩ���������������Ȳ�ķ�Ӧ�� ��ȥ�����ڷ�ʽλ�õ��칹�����ȥ������˳ʽλ�õ���������������˳ʽ������ϩ�ķ�Ӧ�ٶȱȷ�ʽ������ϩ��20����
֬���������ڽ���������Ӧʱ��������������ԭ�ӣ�����ţ����ڷ�ʽλ���������������ұ�������ԭ�Ӻ�����������̼ԭ����ͬһƽ���ϣ���Ӧ�����С� 2.4.1.2 ������������Ӧ���̣�E1���̣� ������������Ӧ���̷��������У���һ������ȥ��������γ�̼�����ӡ��ⲽ�ٶȽ����Ƿ�Ӧ�Ŀ��Ʋ��衣�ڶ���������-�����γ�ϩ���� ���γ�̼�����ӱȽ��ȶ�ʱ����Ӧ���Ȱ�E1���̽��С�
E1��SN1��Ӧ��ͬʱ���������߱�ֵ�������ܼ��ļ��Ժ��¶Ȳ�ͬ���졣�����ܼ����������Ӵ�̼����������ȥ��������E1���̵ķ�Ӧ�� 2.4.2 ��-������Ӧ ��-������Ӧ������ͬ��̼ԭ������������ԭ�ӣ�������γɸ߶Ȼ��õ�ȱ�����ʵ㣨�������ķ�Ӧ��������������ļۼ�״̬�ͻ�ѧ�ṹ�����Է������ֻ�ѧ��Ӧ������������ֵ����������뷴Ӧ��
��������̼-̼˫��������ӳɷ�Ӧ��
�����뷼���廯����Ҳ�ܷ����ӳɷ�Ӧ�����ɻ����������Ժϳɻ�ϩ��
2.4.3 ������ӦӰ������ ��ͬ����Ӧ�����£�������Ӧ����ȡ����Ӧ��ͬʱ�����ġ��������ؽ�������������Ӧ���С� 2.4.3.1 ��Ӧ����ӽṹ��Ӱ�� 1. ��Ӧ����ӵĿռ�ЧӦ ������ԭ��������̼ԭ������֧��ʱ�������˫���ӷ�Ӧ����SN2���̷�Ӧ�����Լ�������-̼ԭ�ӣ�����E2���̷�Ӧ�н�����-��ԭ�ӣ�֧���Ŀռ�ЧӦ��SN2����������Զ�E2����������������ӷ�Ӧ��������SN1��E1�����̣��ڷ�Ӧ�ٶȾ����������γ�ͬ����̼�����ӣ�ֻ�ǵڶ�����ͬ�������н϶�������E1������-����γ�˫���ɼ��ٷ���������ʹ�����ȶ�����SN1ȡ��̼�����������Լ���ϣ����DZ�ѹ������120�����109.5�㣩�������������ӡ��ɼ����۰�˫���ӻ����ӷ�Ӧ����������Ӧ������ 2. ��Ӧ����ӵĵ���ЧӦ ����������-̼ԭ�����������Ի��ţ�X��CN��NO2����������-��ԭ�ӵĻ��ԣ�ʹE2������Ӧ���١� 3. ��ȥ���ŵ����� ��ȥ�����������������ӣ�ʹ��-��ԭ�ӵĵ������ܶ��½���������˫����E2������Ӧ����ȥ���Ŷ���El���̷�Ӧ������Ӱ�졣 2.4.3.2 ��Ӧ������Ӱ�� 1. ���Ӱ�� �Լ��ļ��Զ���˫���ӷ�ӦE2��SN2����Ӱ��ġ����Լ��Ƕ����ӵ�����������Լ���������E2���з�Ӧ�����ס����磬����±�����ˮ�⣬Ϊ����ߴ������ʱ���������Ӧ�����ÿ��Լ�������������Լ�����ΪCH3COO���ļ��Ա�OH�������ö࣬��Ӧ��SN2���̽��С� ��֮����������ȡ��֮�䣬ǿ������������������ȡ��������Ũ�ȵ�ǿ���ڷ����ӻ��ܼ��У�������˫�������̣����Ҷ�E2�ȶ�SN2�����������Ũ�ȵͻ�û�м����ʱ�������ӻ��ܼ��У������ڵ��������̣����Ҷ�SN1�ȶ�E1��Ϊ������ 2. �ܼ������ʣ���Ӱ�� �ܼ��ļ��ԶԷ�ӦӰ������ȡ�������ơ��������ӷ�Ӧ��������̼�����ӣ�����E1��SN1���̽��У������ܼ�����ֻ�ٽ������м�������ʣ�����E1��SNl����ı���Ӱ���С�� ��˫���ӷ�Ӧ���ڼ���С���ܼ��ж����γ�E2�Ĺ���̬����������������ϩ�����ɡ� 3. �¶ȵ�Ӱ�� ��Ӧ�����ǵ����ӻ���˫�������̣���߷�Ӧ�¶ȶ�������������Ӧ�Ľ��С��������������Ӧ�Ļ������Ҫ������-̼�����ԭ�� 2.5 �������Ӧ �������Ӧ�ֳ����ɻ���Ӧ���Ǿ�ϸ�л��ϳ���һ�����Ҫ�ķ�Ӧ����һ��������ͨ�����ܺܿ������ȥ���ǿ���������Ӧ������ӦҲ���ܵ�ijЩ���ʵ����ƣ���Щ�����ܷdz�������������ϣ�ʹ��Ӧ��ֹ��Ϊ��ʹ�������Ӧ�ܹ�˳�����������Ȳ���һ������������������õķ��������֣�����ⷨ������ⷨ�͵���ת�Ʒ��� 2.5.1 ����ⷨ ���������ȵ�һ���¶ȷ�������⣬�������������ͬ�����������������¶Ȳ�ͬ�����磬�ȷ��ӵ��������100�����Ͽɾ���һ�����ٶȣ����������ѡ�ȩ��ͪ���ȵ�800~1000��ʱ��⣻�����л������������¶ȵ�Щ���ļ�Ǧ����ͨ��������600���ʯӢ�ܣ������ɼ��������
���������Ļ������ѽ������¶ȵ�Щ�����纬��O-O���Ĺ���������������ż��˫�춡�涼�dz��õ���������
2.5.2 ����ⷨ �����ܵ������������������Ӿ��нϸߵ����������ǿ������㻯ѧ����������Ҫ�����������磬±�ط����ù������������ǵ�ԭ�ӣ����������κ��¶������У�������ͨ�����ڹ������ǿ�ȿ���������������ٶȡ�
2.5.3 ����ת�Ʒ� �ؽ������Ӿ��е�ʧ���ӵ����ܳ������ڴ�ijЩ��������ķֽ⡣���磬�������ӽ�һ������ת�Ƹ���������ʹ������һ���ǻ��������һ�����ȶ����ǻ������ӡ����۵������ӿ��Դӹ������嶡���л�ȡһ�����ӣ�ʹ�������嶡��ת���һ�������������һ�����ӡ�
�������Ӧ����������Ӧ���䷴Ӧ���̰��������Σ����������������Ĵ��ݺ�������ֹ���������Ӧ�ھ�ϸ�л��ϳ����й㷺��Ӧ�á� 2.6 �ӳɷ�Ӧ �ӳɷ�Ӧ���������ͣ�����ӳɡ��˼ӳɺ�������ӳɡ� 2.6.1 ��ӳ� ��ӳ�һ�㷢����̼-̼˫���ϣ���Ϊϩ����Ȳ�������е���-����ֻ�нϴ�Ļ�ԣ����ֳ����ԣ���������������������Լ�������ӳɷ�Ӧ�����õ����Լ��У�ǿ�ᣨ�������ᡢ��±�ᣩ��Lewis�ᣨ����FeCl3��A1C13��HgC12����±�ء���±�ᡢ±���顢��������������������ȡ��䷴Ӧ���̷��������У���������̼�������м����������ʿ��Ʋ��衣
Ȼ���ǣ�
ϩ���Ľṹ��ͬ���Է�Ӧ�ٶȵ�Ӱ��Ҳ��ȫ��������ֲ��ӳ�����һ�¡���̼-̼˫�������и�����ȡ����ʱ������������̼-̼˫���ϵĵ������ܶȿ�ʹ̼�������ȶ�������ӿ��˷�Ӧ�ٶȡ�������������ȡ����ʱ�����ڽ�����̼-̼˫���ϵĵ������ܶȣ�������̼�����ӵ��ȶ��ԣ���������˷�Ӧ�ٶȡ� 2.6.2 �˼ӳ� �˼ӳ�������Ҫ����̼��˫����̼�������˼ӳɡ���̼��˫������ԭ�ӵĵ縺�Ա�̼ԭ�Ӹߵö࣬�����ԭ�ڴ��в��ָ���ɣ���̼ԭ������в�������ɡ�
̼��˫���ڽ��мӳɷ�Ӧʱ��������ɵ�������Ҫ�ȴ�����ɵ�̼ԭ���ȶ��ö࣬����ڼ��Դ��������£����Ǵ�����ɵ�̼ԭ�������Լ�������Ӧ����̼��˫�����ڷ����˼ӳɷ�Ӧ�� 2.7 ���ŷ�Ӧ ���ŷ�Ӧ��ָ���Լ������»�������Ӱ���£��л�������з���ijЩ�����ת�ƣ��γ���һ�ֻ�����ķ�Ӧ�����ŷ�Ӧ����ֻܶ࣬���۷����廯�����������Ǩ�ƣ��Լ�Ǩ�Ʒ������ڽ�����ԭ�Ӽ��1,2-Ǩ�ơ�
2.7.1 ���Ӽ����� ���Ӽ����ŷ�Ӧ�������ܹ���÷��ѳ�����Ǩ�ƻ���Z�������磬���������N-���������������ŷ�Ӧ��
����ͨ���û������ȷ��ӣ������ȷ�������������������ȡ���� 2.7.2 ������������ 2.7.2.1 ���������� ����������������⻯ż����ת�����������ķ�Ӧ��
2.7.2.2 N-ȡ������������ N-ȡ������������������Ǩ�ƻ��ӵ�ԭ��Ǩ�Ƶ�������λ���λ�ϡ�������������Ǩ�ƣ��������Ե����ŷ�Ӧ���ٷ�����N-�����������������ᴦ��ʱ�������ţ���Ҫ���ɶ�λ�칹���
2.7.2.3 �ǻ���Ǩ�� Ǩ�ƻ�����Ϊ�����ʵ������ԭ����֧�����ϵĵ��Ӷԣ���֧��Ǩ���������ϣ��������ų�Ϊ�����������š����磬��ϡ���������ڱ����ǰ���������OH����Ǩ�ƣ������˰����ӡ�
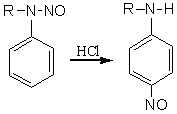
|